Preformulation Study of Electrospun Haemanthamine-Loaded Amphiphilic Nanofibers Intended for a Solid Template for Self-Assembled Liposomes

 ,
,  ,
,  ,
,  and
and
Abstract
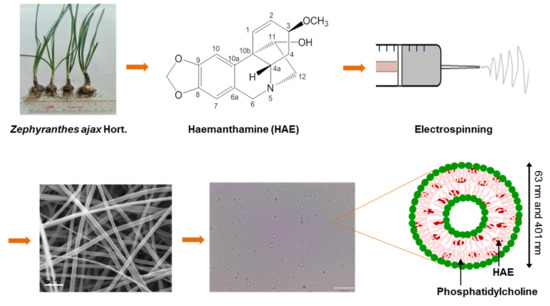
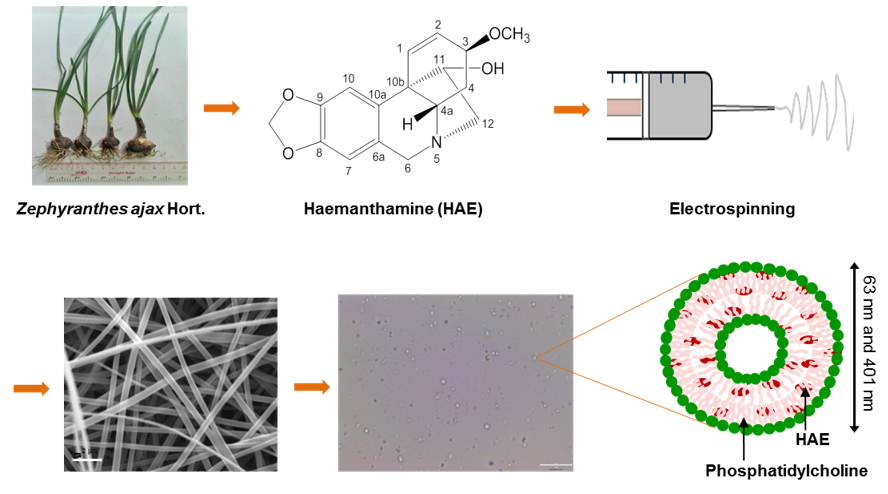{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
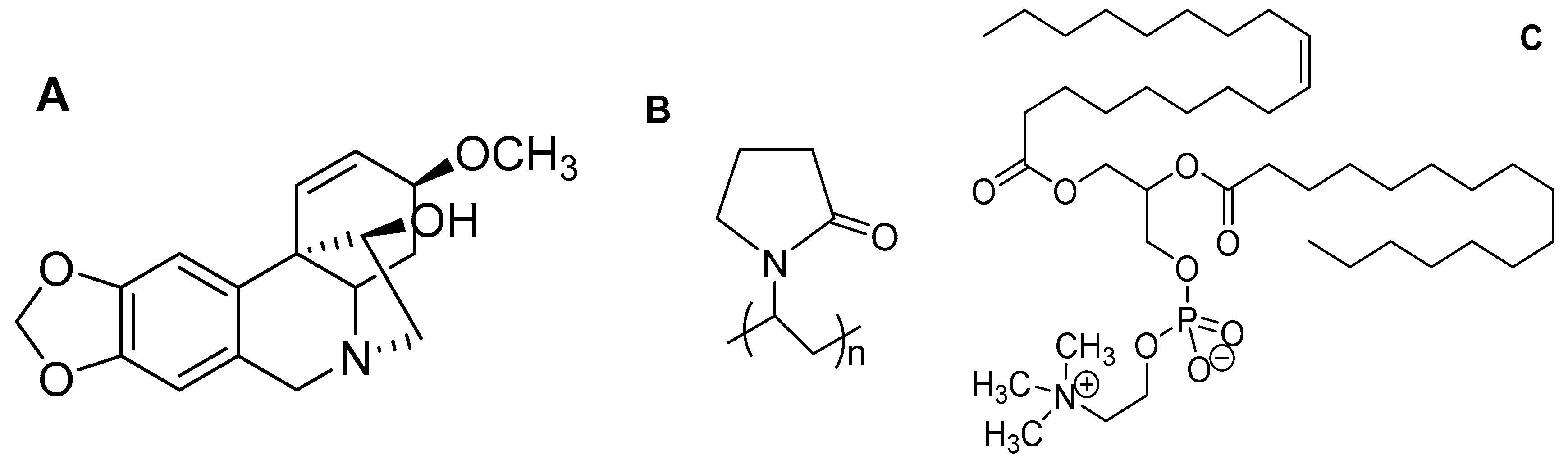
2.1. Materials
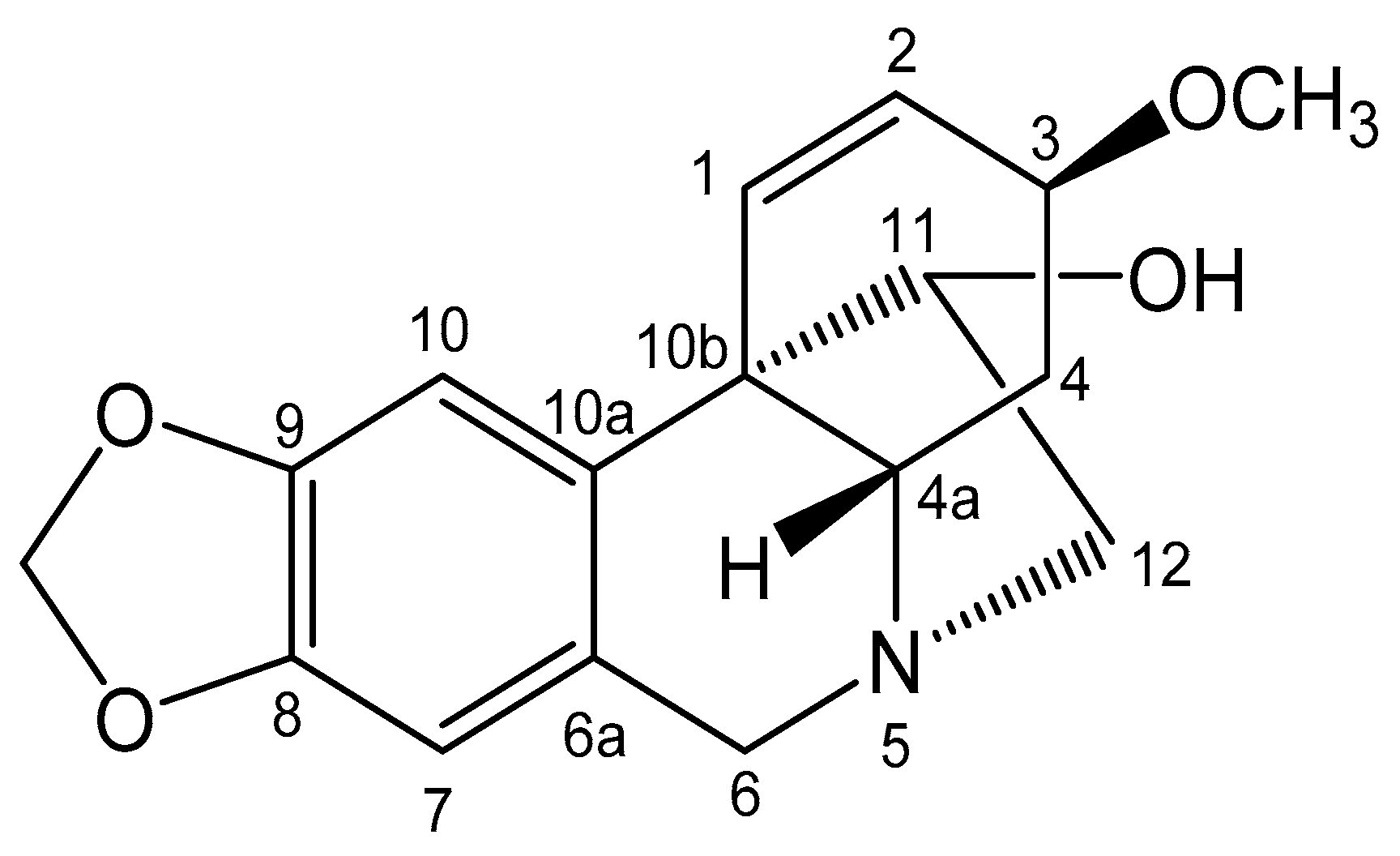
2.2. NMR Structure Analysis
2.3. Fabrication of Amphiphilic Nanofibers
2.4. The Geometric Properties and Surface Morphology of NFs
2.5. Optical Microscopy of Self-Assembled Liposomes
2.6. Photon Correlation Spectroscopy (PCS)
2.7. Fourier Transform Infrared Spectroscopy
2.8. X-ray Powder Diffraction (XRPD)
2.9. Differential Scanning Calorimetry (DSC)
2.10. In Vitro Drug Release
3. Results and Discussion
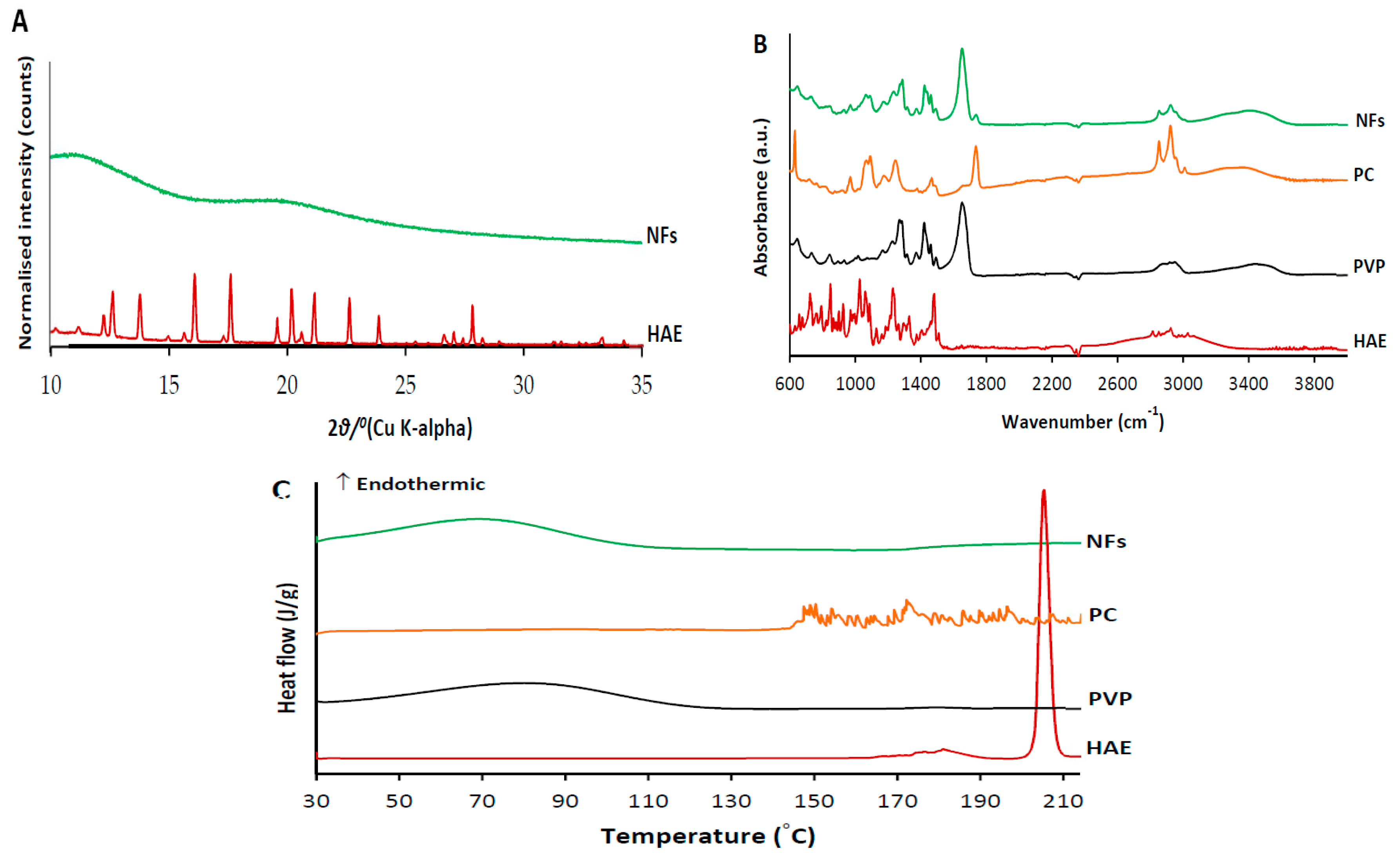
3.1. NMR Spectroscopy Analysis of HAE
3.2. SEM Analysis of Amphiphilic NFs and Templates
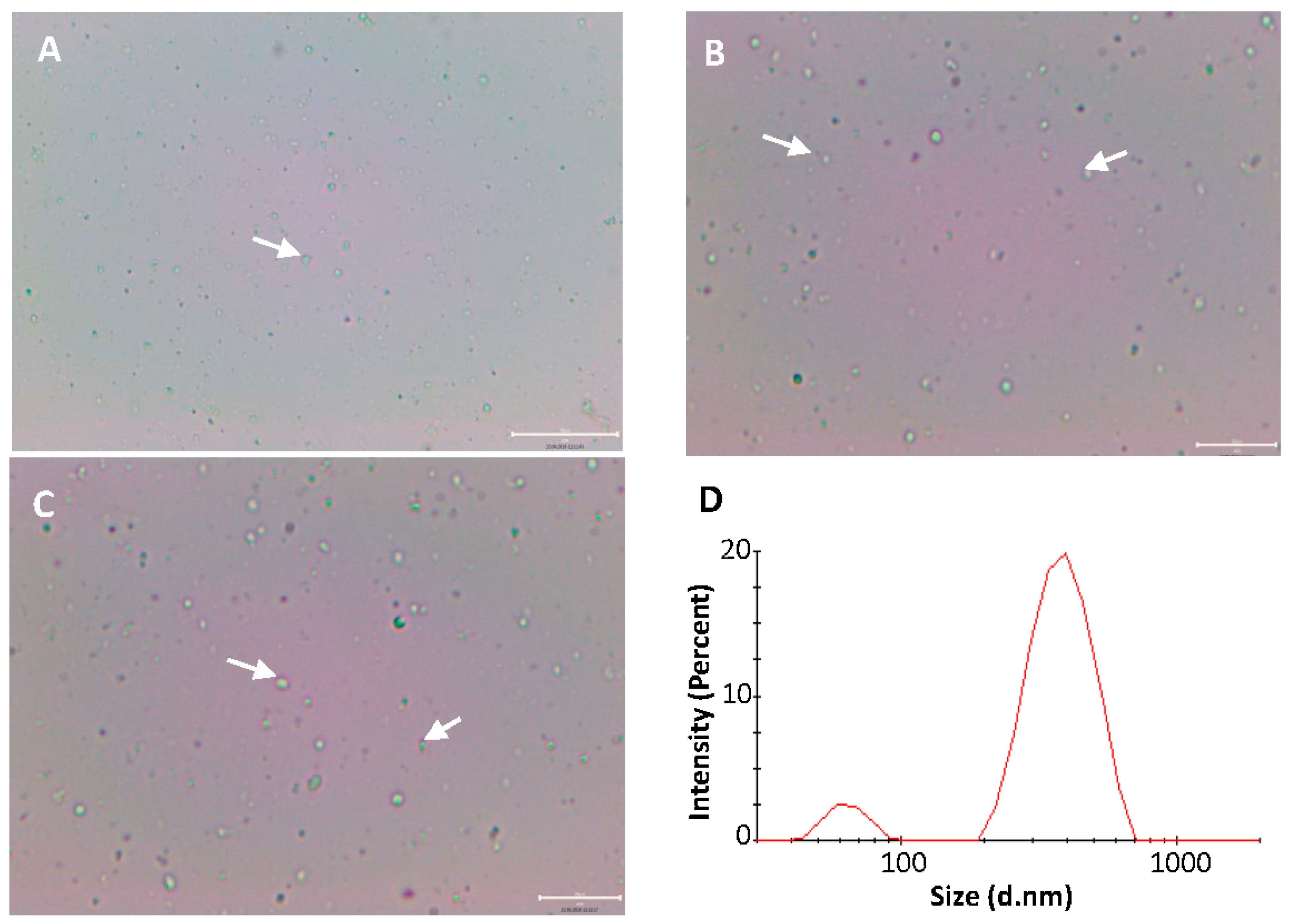
3.3. Optical Microscopy of Self-Assembled Liposomes
3.4. Particle Size Analysis of Self-Assembled Liposomes
3.5. Physical Solid-State Properties
3.6. In Vitro Drug Release
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yurteri, C.R.; Hartman, P.A.; Marijnissen, J.C.M. Producing pharmaceutical particles via electrospraying with an emphasis on nano and ano structured particles—A Review. KONA Powder Part. J. 2010, 28, 91–115. [Google Scholar] [CrossRef]
- Havelek, R.S.; Kralovec, K.; Bruckova, L.; Cahlikova, L.; Rezacova, M.; Opletal, L.; Bilkova, Z. Effect of Amaryllidaceae alkaloids haemanthamine and haemanthidine on cell viability, apoptosis and cell cycle progression in human T-lymphoblast cell line. FEBS J. 2013, 280, 322–323. [Google Scholar]
- Van Goietsenoven, G.; Andolfi, A.; Lallemand, B.; Cimmino, A.; Lamoral-Theys, D.; Gras, T.; Abou-Donia, A.; Dubois, J.; Lefranc, F.; Mathieu, V.; et al. Amaryllidaceae alkaloids belonging to different structural subgroups display activity against apoptosis-resistant cancer cells. J. Nat. Prod. 2010, 73, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.J.; Bastida, J.; Viladomat, F.; van Staden, J. Cytotoxic agents of the crinane series of amaryllidaceae alkaloids. Nat. Prod. Commun. 2012, 7, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Weniger, B.; Italiano, L.; Beck, J.P.; Bastida, J.; Bergoñon, S.; Codina, C.; Lobstein, A.; Anton, R. Cytotoxic Activity of Amaryllidaceae alkaloids. Planta Med. 1995, 61, 77–79. [Google Scholar] [CrossRef]
- Pellegrino, S.; Meyer, M.; Zorbas, C.; Bouchta, S.A.; Saraf, K.; Pelly, S.C.; Yusupova, G.; Evidente, A.; Mathieu, V.; Kornienko, A.; et al. The Amaryllidaceae alkaloid haemanthamine binds the eukaryotic ribosome to repress cancer cell growth. Structure 2018, 26, 416–425.e4. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Yu, D.G.; Branford-White, C.; Williams, G.R.; Bligh, S.A.; White, K.; Zhu, L.M.; Chatterton, N.P. Self-assembled liposomes from amphiphilic electrospun nanofibers. J. Soft Matter 2011, 7, 8239–8247. [Google Scholar] [CrossRef]
- Sebe, I.; Szabó, P.; Kállai-Szabó, B.; Zelkó, R. Incorporating small molecules or biologics into nanofibers for optimized drug release: A Review. Int. J. Pharm. 2015, 494, 516–530. [Google Scholar] [CrossRef]
- He, D.; Hu, B.; Yao, Q.F.; Wang, K.; Yu, S.H. Large-scale synthesis of flexible free-standing SERS substrates with high sensitivity: Electrospun PVA nanofibers embedded with controlled alignment of silver nanoparticles. ACS Nano 2009, 3, 3993–4002. [Google Scholar] [CrossRef]
- Lu, X.; Wang, C.; Wei, Y. Synthesis by electrospinning and their applications. One-dimensional composite nanomaterials. Small 2009, 5, 2349–2370. [Google Scholar] [CrossRef] [PubMed]
- Paaver, U.; Heinämäki, J.; Laidmäe, I.; Lust, A.; Kozlova, J.; Sillaste, E.; Kirsimäe, K.; Veski, P.; Kogermann, K. Electrospun nanofibers as a potential controlled-release solid dispersion system for poorly water-soluble drugs. Int. J. Pharm. 2015, 479, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.N.; Laidmäe, I.; Kogermann, K.; Lust, A.; Meos, A.; Nguyen, C.N.; Heinämäki, J. Development of electrosprayed artesunate-loaded core-shell nanoparticles. Drug Dev. Ind. Pharm. 2017, 43, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Ingebrigtsen, L.; Brandl, M. Determination of the size distribution of liposomes by SEC fractionation, and PCS analysis and enzymatic assay of lipid content. AAPS PharmSciTech 2002, 3, 9–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lopez, S.; Bastida, J.; Viladomat, F.; Codina, C. Solid-phase extraction and reversed-phase high-performance liquid chromatography of the five major alkaloids in Narcissus confusus. Phytochem. Anal. 2002, 13, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.; Viladomat, F.; Llabres, J.M.; Codina, C.; Feliz, M.; Rubiralta, M. Alkaloids from Narcissus confuses. Phytochemistry 1987, 26, 1519–1524. [Google Scholar] [CrossRef]
- Bohno, M.; Sugie, K.; Imase, H.; Yusof, Y.B.; Oishi, T.; Chida, N. Total synthesis of Amaryllidaceae alkaloids, (+)-vittatine and (+)-haemanthamine, starting from d-glucose. Tetrahedron 2007, 63, 6977–6989. [Google Scholar] [CrossRef]
- Kogermann, K.; Penkina, A.; Predbannikova, K.; Jeeger, K.; Veski, P.; Rantanen, J.; Naelapää, K. Dissolution testing of amorphous solid dispersions. Int. J. Pharm. 2013, 444, 40–46. [Google Scholar] [CrossRef]
- Shi, N.Q.; Yao, J.; Wu, Y.; Wang, X.L. Effect of polymers and media type on extending the dissolution of amorphous pioglitazone and inhibiting the recrystallization from a supersaturated state. Drug Dev. Ind. Pharm. 2014, 40, 1112–1122. [Google Scholar] [CrossRef]
- Li, Z.; Wang, C. Effects of working parameters on electrospinning. In One-Dimensional Nanostructures; Springer: Berlin/Heidelberg, Germany, 2013; pp. 15–28. [Google Scholar]
- Hu, X.; Liu, S.; Zhou, G.; Huang, Y.; Xie, Z.; Jing, X. Electrospinning of polymeric nanofibers for drug delivery applications. J. Control. Release 2014, 185, 12–21. [Google Scholar] [CrossRef]
- Tantipolphan, R.; Rades, T.; McQuillan, A.J.; Medlicott, N.J. Adsorption of bovine serum albumin (BSA) onto lecithin studied by attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. Int. J. Pharm. 2007, 337, 40–47. [Google Scholar] [CrossRef] [PubMed]
- PharmacyCodes. Haemanthamine [Online]. Available online: https://pharmacycode.com/Haemanthamine.html (accessed on 20 August 2019).
- Maiti, K.; Mukherjee, K.; Gantait, A.; Saha, B.P.; Mukherjee, P.K. Curcumin-phospholipid complex: Preparation, therapeutic evaluation and pharmacokinetic study in rats. Int. J. Pharm. 2007, 330, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Koynova, R.; Caffrey, M. Phases and phase transitions of the phosphatidylcholines. Biochim. Biophys. Acta 1998, 1376, 91–145. [Google Scholar] [CrossRef]
- Khan, A.U.R.; Nadeem, M.; Bhutto, M.A.; Yu, F.; Xie, X.; El-Hamshary, H.; El-Faham, A.; Ibrahim, U.A.; Mo, X. Physico-chemical and biological evaluation of PLCL/SF nanofibers loaded with oregano essential oil. Pharmaceutics 2019, 11, 386. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viet Nguyen, K.; Laidmäe, I.; Kogermann, K.; Lust, A.; Meos, A.; Viet Ho, D.; Raal, A.; Heinämäki, J.; Thi Nguyen, H. Preformulation Study of Electrospun Haemanthamine-Loaded Amphiphilic Nanofibers Intended for a Solid Template for Self-Assembled Liposomes. Pharmaceutics 2019, 11, 499. https://doi.org/10.3390/pharmaceutics11100499
Viet Nguyen K, Laidmäe I, Kogermann K, Lust A, Meos A, Viet Ho D, Raal A, Heinämäki J, Thi Nguyen H. Preformulation Study of Electrospun Haemanthamine-Loaded Amphiphilic Nanofibers Intended for a Solid Template for Self-Assembled Liposomes. Pharmaceutics. 2019; 11(10):499. https://doi.org/10.3390/pharmaceutics11100499
Chicago/Turabian StyleViet Nguyen, Khan, Ivo Laidmäe, Karin Kogermann, Andres Lust, Andres Meos, Duc Viet Ho, Ain Raal, Jyrki Heinämäki, and Hoai Thi Nguyen. 2019. "Preformulation Study of Electrospun Haemanthamine-Loaded Amphiphilic Nanofibers Intended for a Solid Template for Self-Assembled Liposomes" Pharmaceutics 11, no. 10: 499. https://doi.org/10.3390/pharmaceutics11100499
APA StyleViet Nguyen, K., Laidmäe, I., Kogermann, K., Lust, A., Meos, A., Viet Ho, D., Raal, A., Heinämäki, J., & Thi Nguyen, H. (2019). Preformulation Study of Electrospun Haemanthamine-Loaded Amphiphilic Nanofibers Intended for a Solid Template for Self-Assembled Liposomes. Pharmaceutics, 11(10), 499. https://doi.org/10.3390/pharmaceutics11100499