Quasi-Dynamic Dissolution of Electrospun Polymeric Nanofibers Loaded with Piroxicam


 , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Fiber Mats
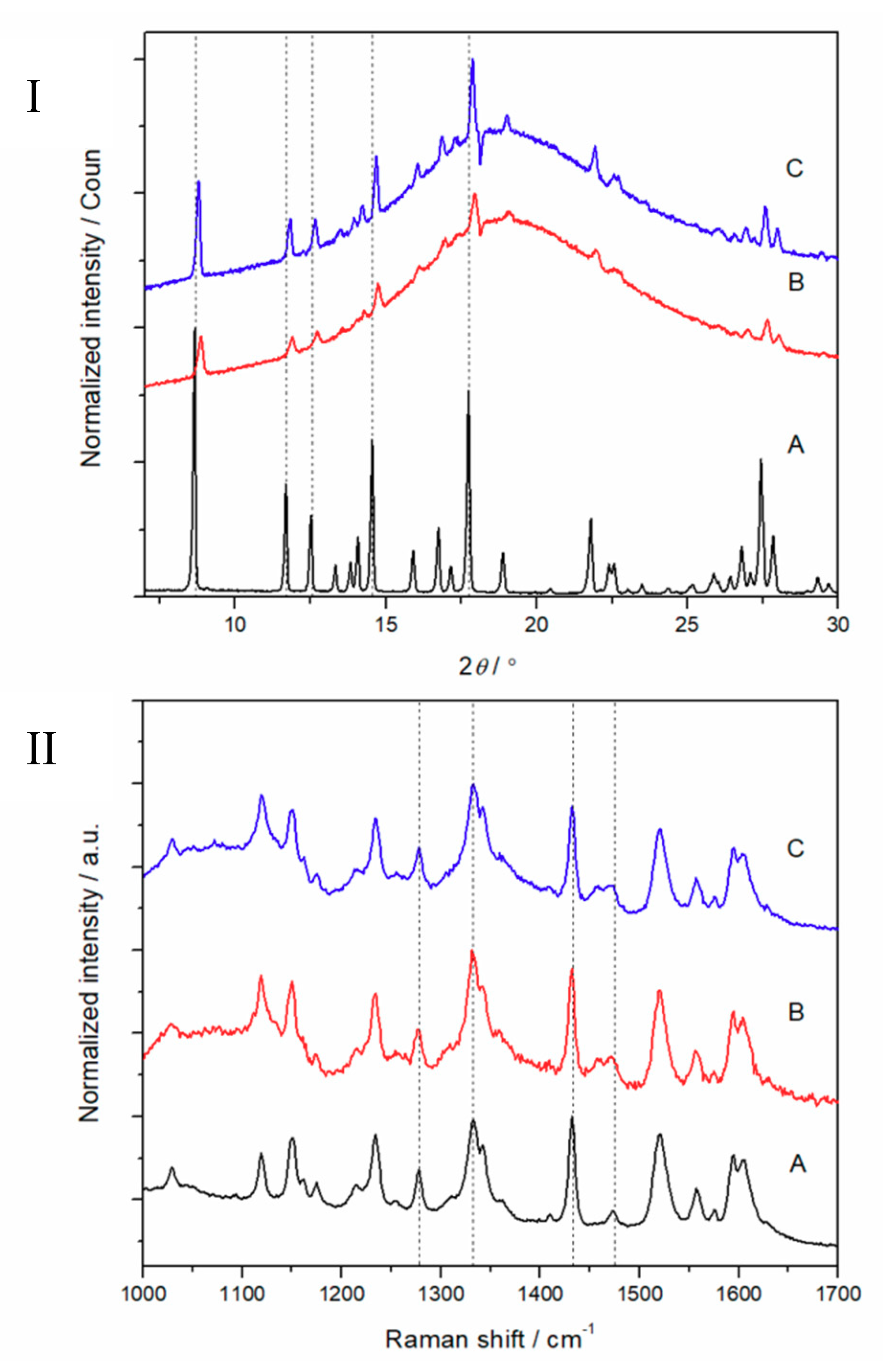
2.3. Physical Appearance and Solid-State Characterization
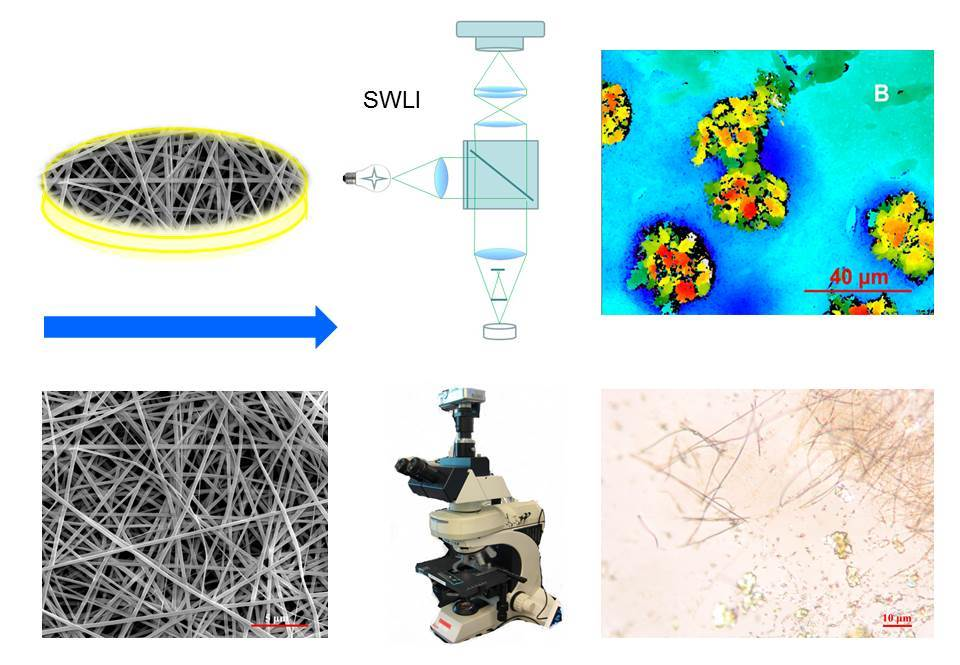
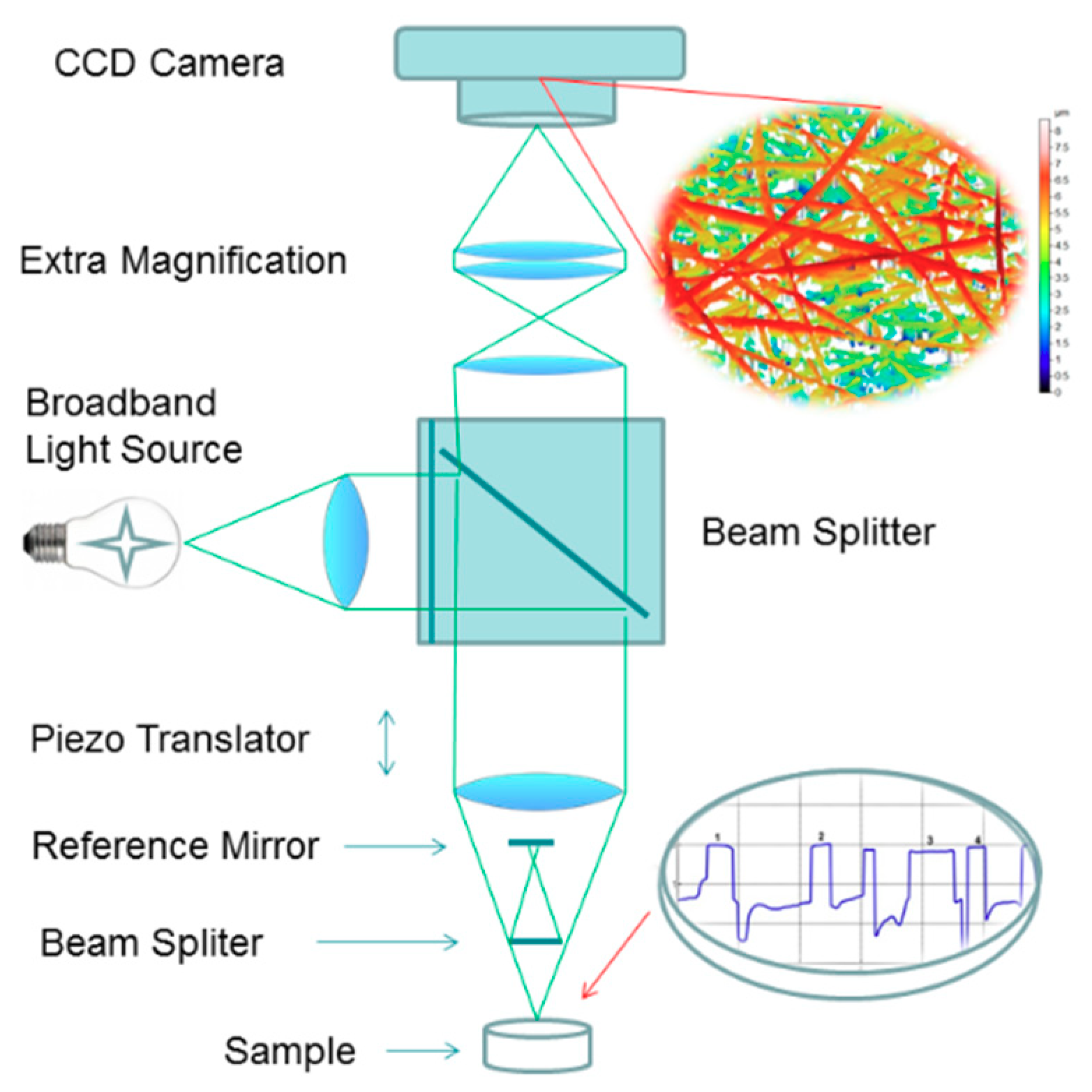
2.4. In Situ Wetting and Dissolution Tests of Fibers
2.5. Dissolution Test
3. Results and Discussion
3.1. Physical Appearance of Nanofibers
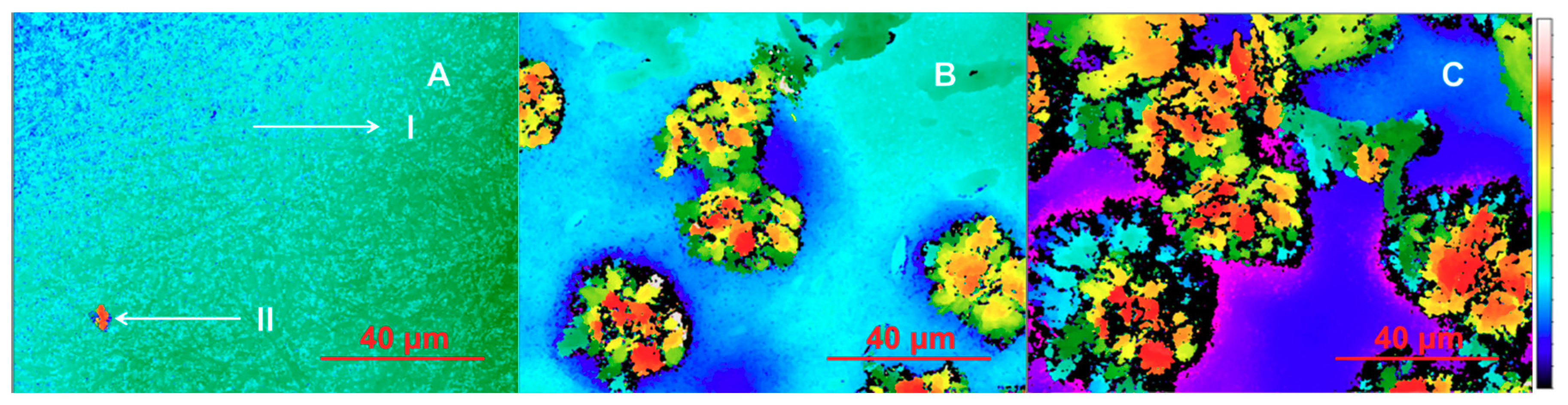
3.2. Physical Solid-State Properties of Nanofibers
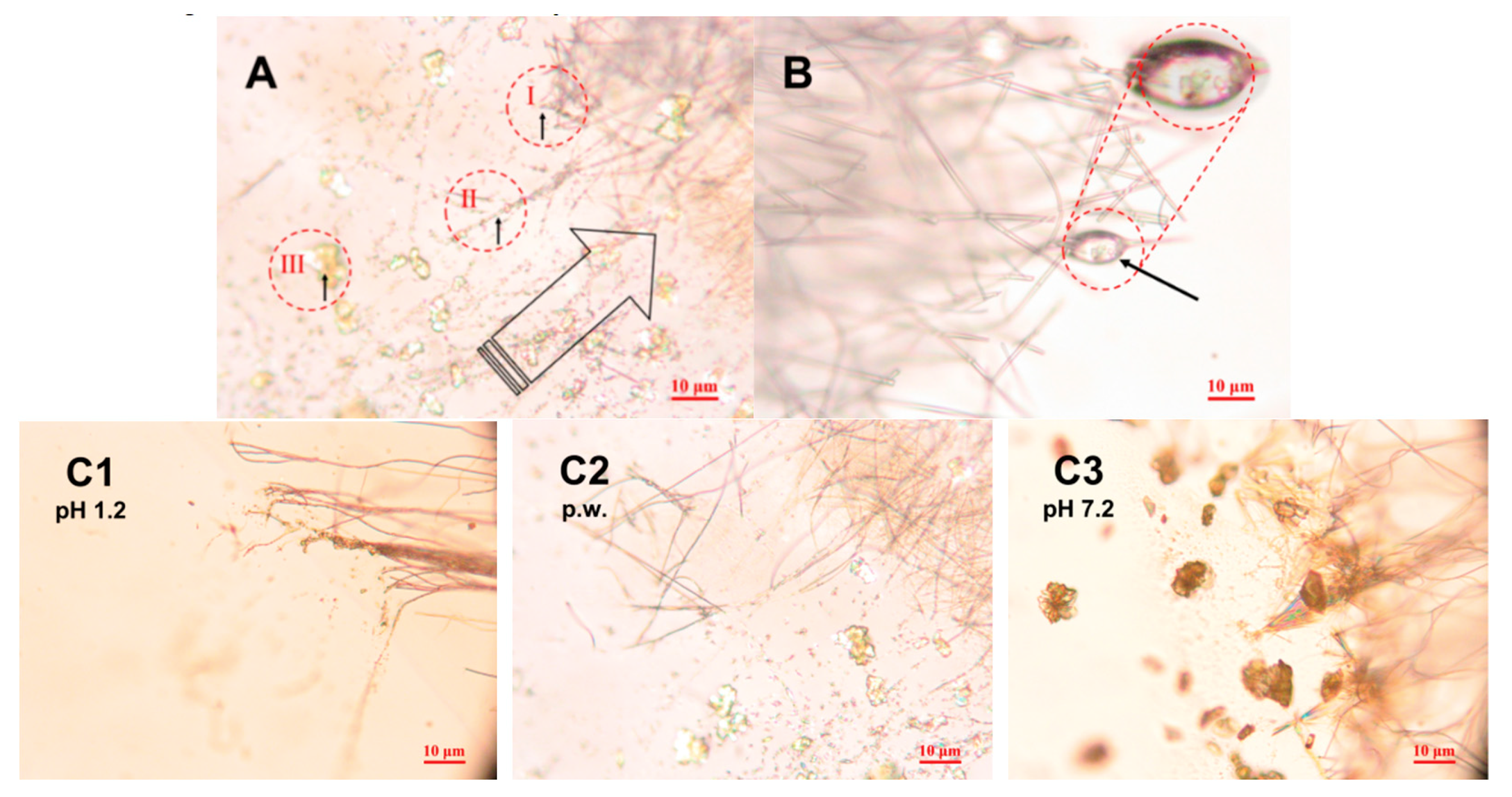
3.3. In Situ Wetting and Initial Dissolution of Fibers
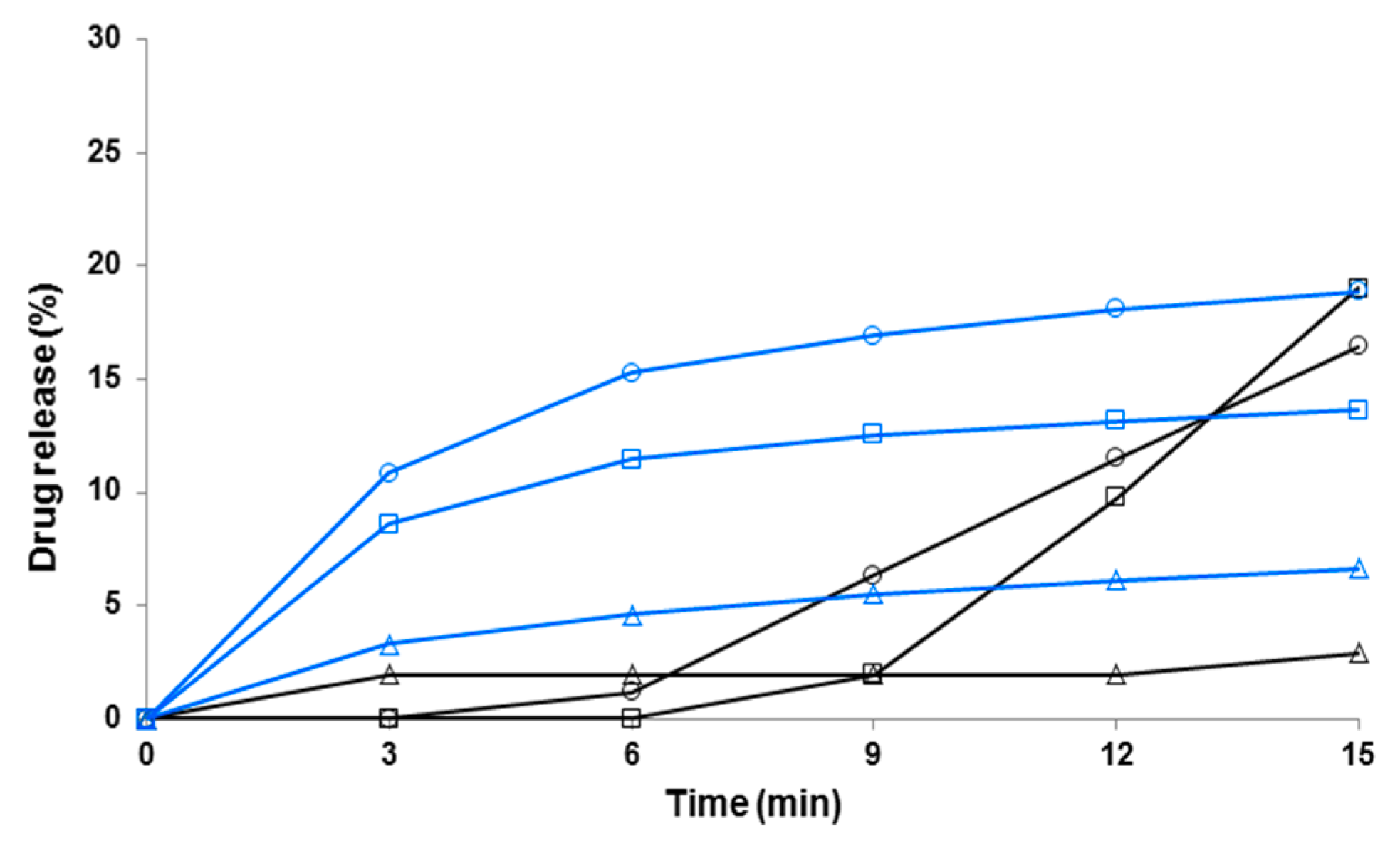
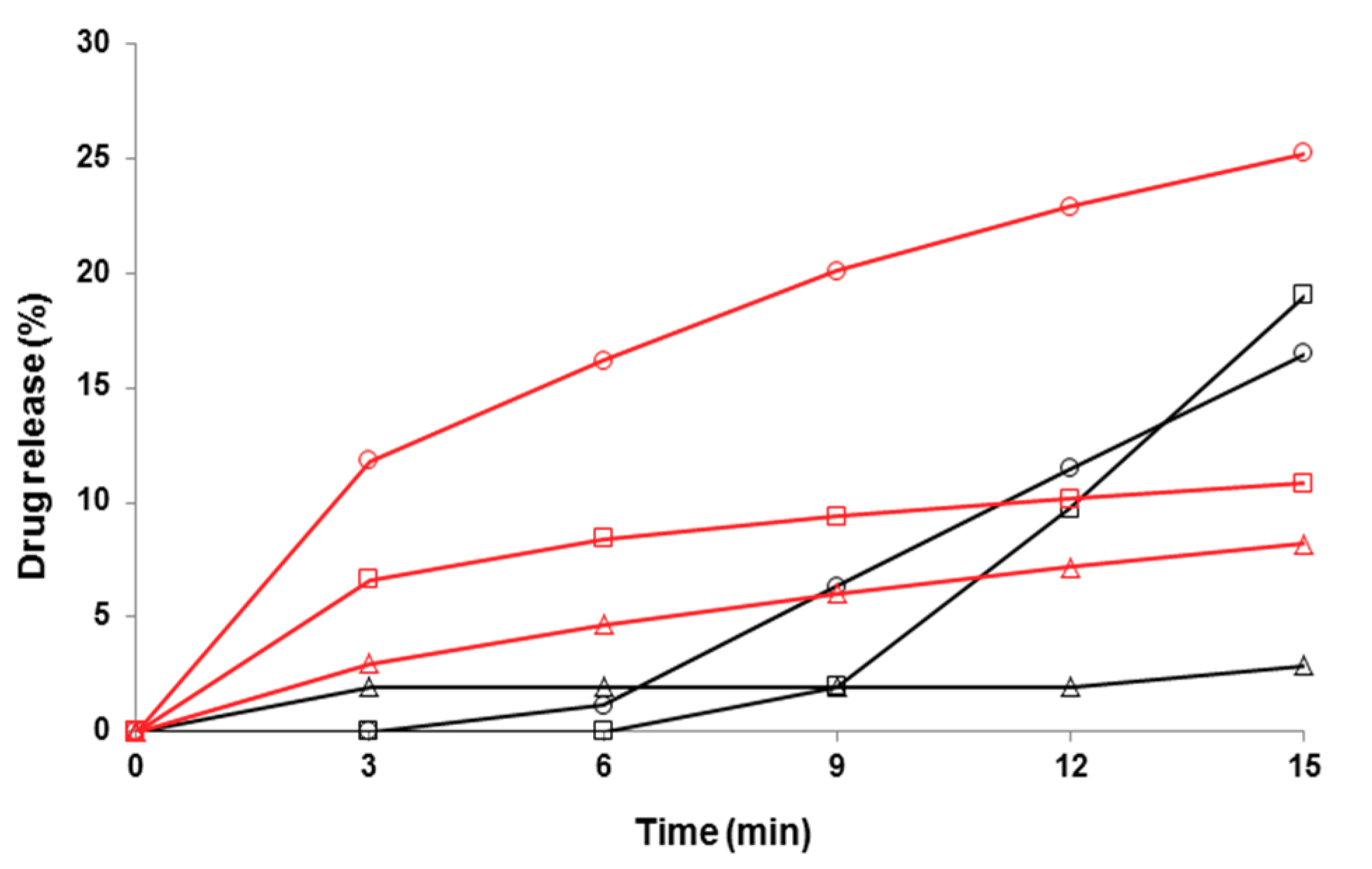
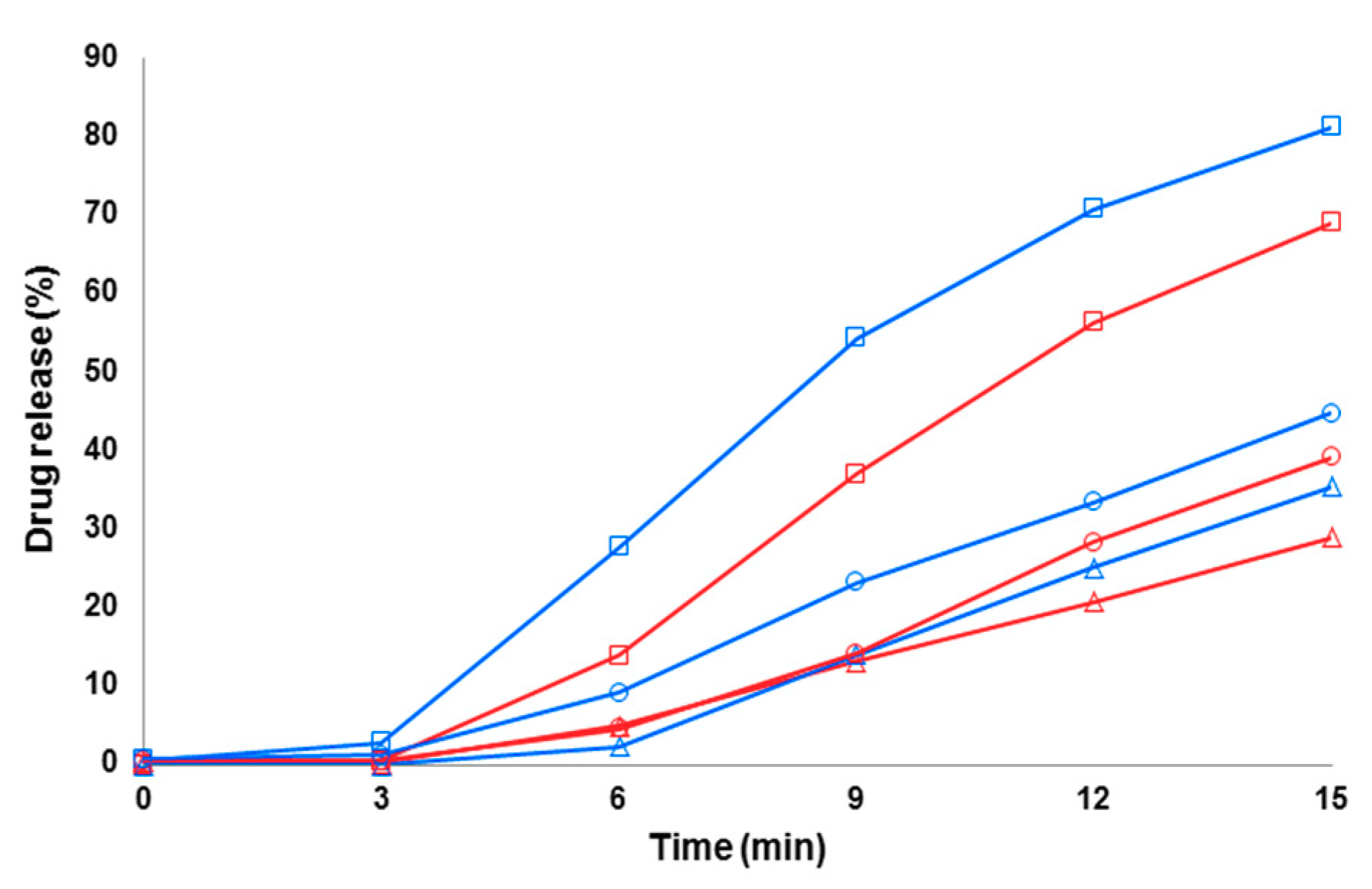
3.4. Dissolution of Fibers
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rošic, R.; Kocbek, P.; Pelipenko, J.; Kristl, J.; Baumgartner, S. Nanofibers and their biomedical use. Acta Pharm. 2013, 63, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, J.D.; Schauer, C.L. A Review: Electrospinning of biopolymer nanofibers and their applications. Polym. Rev. 2008, 48, 317–352. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Kundu, S.C. Electrospinning: A fascinating fiber fabrication technique. Biotechnol. Adv. 2010, 28, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Rošic, R.; Pelipenko, J.; Kocbek, P.; Baumgartner, S.; Bešter-Rogač, M.; Kristl, J. The role of rheology of polymer solutions in predicting nanofiber formation by electrospinning. Eur. Polym. J. 2012, 48, 1374–1384. [Google Scholar] [CrossRef]
- Pelipenko, J.; Kocbek, P.; Kristl, J. Critical attributes of nanofibers: Preparation, drug loading, and tissue regeneration. Int. J. Pharm. 2015, 484, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Sebe, I.; Szabo, P.; Kallai-Szabo, B.; Zelko, R. Incorporating small molecules or biologics into nanofibers for optimized drug release: A review. Int. J. Pharm. 2015, 494, 516–530. [Google Scholar] [CrossRef]
- Torres-Martinez, E.J.; Cornejo Bravo, J.M.; Serrano Medina, A.; Pérez González, G.L.; Villarreal Gómez, L.J. A Summary of Electrospun Nanofibers as Drug Delivery System: Drugs Loaded and Biopolymers Used as Matrices. Curr. Drug Deliv. 2018, 15, 1360–1374. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Thakkar, S.; Misra, M. Electrospun polymeric nanofibers: New horizons in drug delivery. Eur. J. Pharm. Sci. 2017, 107, 148–167. [Google Scholar] [CrossRef]
- Haider, A.; Haider, S.; Kang, I.K. A comprehensive review summarizing the effect of electrospinning parameters and potential applications of nanofibers in biomedical and biotechnology. Arab. J. Chem. 2018, 11, 1165–1188. [Google Scholar] [CrossRef]
- Lim, C.T. Nanofiber technology: Current status and emerging developments. Prog. Polym. Sci. 2017, 70, 1–17. [Google Scholar]
- Wang, J.; Windbergs, M. Functional electrospun fibers for the treatment of human skin wounds. Eur. J. Pharm. Biopharm. 2017, 119, 283–299. [Google Scholar] [CrossRef]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and Electrospun Nanofibers: Methods, Materials, and Applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef]
- Hu, X.; Liu, S.; Zhou, G.; Huang, Y.; Xie, Z.; Jing, X. Electrospinning of polymeric nanofibers for drug delivery applications. J. Control. Release 2014, 185, 12–21. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.; Xiang, D. Nanoparticle drug delivery systems: An excellent carrier for tumor peptide vaccines. Drug Deliv. 2018, 25, 1319–1327. [Google Scholar] [CrossRef]
- Paaver, U.; Heinämäki, J.; Laidmäe, I.; Lust, A.; Kozlova, J.; Sillaste, E.; Kirsimäe, K.; Veski, P.; Kogermann, K. Electrospun nanofibers as a potential controlled-release solid dispersion system for poorly water-soluble drugs. Int. J. Pharm. 2015, 479, 252–260. [Google Scholar] [CrossRef]
- Paaver, U.; Heinämäki, J.; Kassamakov, I.; Haeggstrom, E.; Ylitalo, T.; Nolvi, A.; Kozlova, J.; Laidmäe, I.; Kogermann, K.; Veski, P. Nanometer depth resolution in 3D topographic analysis of drug-loaded nanofibrous mats without sample preparation. Int. J. Pharm. 2014, 462, 29–37. [Google Scholar] [CrossRef]
- Sandler, N.; Kassamakov, I.; Ehlers, H.; Genina, N.; Ylitalo, T.; Haeggstrom, E. Rapid interferometric imaging of printed drug laden multilayer structures. Sci. Rep. 2014, 4, 4020. [Google Scholar] [CrossRef]
- Genina, N.; Fors, D.; Vakili, H.; Ihalainen, P.; Pohjala, L.; Ehlers, H.; Kassamakov, I.; Haeggstrom, E.; Vuorela, P.; Peltonen, J.; et al. Tailoring controlled-release oral dosage forms by combining inkjet and flexographic printing techniques. Eur. J. Pharm. Sci. 2012, 47, 615–623. [Google Scholar] [CrossRef]
- Hanhijärvi, K.; Majava, T.; Kassamakov, I.; Heinämäki, J.; Aaltonen, J.; Haapalainen, J.; Haeggström, E.; Yliruusi, J. Scratch resistance of plasticized hydroxypropyl methylcellulose (HPMC) films intended for tablet coatings. Eur. J. Pharm. Biopharm. 2010, 74, 371–376. [Google Scholar] [CrossRef]
- O’Bryan, C.S.; Bhattacharjee, T.; Hart, S.; Kabb, C.P.; Schulze, K.D.; Chilakala, I.; Sumerlin, B.S.; Sawyer, W.G.; Angelini, T.E. Self-assembled micro-organogels for 3D printing silicone structures. Sci. Adv. 2017, 3, e1602800. [Google Scholar] [CrossRef]
- Wickström, H.; Hilgert, E.; Nyman, O.J.; Desai, D.; Şen Karaman, D.; De Beer, T.; Sandler, N.; Rosenholm, M.J. Inkjet Printing of Drug-Loaded Mesoporous Silica Nanoparticles—A Platform for Drug Development. Molecules 2017, 22, 2020. [Google Scholar] [CrossRef]
- Florek, C.A.; Kohn, J.B.; Michniak-Kohn, B.B.; Thakur, R.A. Electrospun Matrices for Delivery of Hydrophilic and Lidophilic Compounds. Available online: https://patentsgooglecom/patent/EP2079416A2/en (accessed on 9 August 2019).
- Baier, S.; Given, P.; Kanjanapongkul, K.; Weiss, J. Formation of Conjugated Protein by Electrospinning. Available online: https://patentsgooglecom/patent/WO2013151694A1/de (accessed on 9 August 2019).
- Kassamakov, I.V.; Seppänen, H.O.; Oinonen, M.J.; Hæggström, E.O.; Österberg, J.M.; Aaltonen, J.P.; Saarikko, H.; Radivojevic, Z.P. Scanning white light interferometry in quality control of single-point tape automated bonding. Microelectron. Eng. 2007, 84, 114–123. [Google Scholar] [CrossRef]
- Kassamakov, I.; Hanhijärvi, K.; Abbadi, I.; Aaltonen, J.; Ludvigsen, H.; Haeggström, E. Scanning white-light interferometry with a supercontinuum source. Opt. Lett. 2009, 34, 1582–1584. [Google Scholar] [CrossRef]
- Kogermann, K.; Veski, P.; Rantanen, J.; Naelapää, K. X-ray powder diffractometry in combination with principal component analysis—A tool for monitoring solid state changes. Eur. J. Pharm. Sci. 2011, 43, 278–289. [Google Scholar] [CrossRef]
- Brewster, M.E.; Verreck, G.; Chun, I.; Rosenblatt, J.; Mensch, J.; Van Dijck, A.; Noppe, M.; Ariën, A.; Bruining, M.; Peeters, J. The use of polymer-based electrospun nanofibers containing amorphous drug dispersions for the delivery of poorly water-soluble pharmaceuticals. Pharmazie 2004, 59, 387–391. [Google Scholar]
- Verreck, G.; Chun, I.; Rosenblatt, J.; Peeters, J.; Dijck, A.V.; Mensch, J.; Noppe, M.; Brewster, M.E. Incorporation of drugs in an amorphous state into electrospun nanofibers composed of a water-insoluble, nonbiodegradable polymer. J. Control. Release 2003, 92, 349–360. [Google Scholar] [CrossRef]
- Zupančič, Š.; Preem, L.; Kristl, J.; Putrinš, M.; Tenson, T.; Kocbek, P.; Kogermann, K. Impact of PCL nanofiber mat structural properties on hydrophilic drug release and antibacterial activity on periodontal pathogens. Eur. J. Pharm. Sci. 2018, 122, 347–358. [Google Scholar] [CrossRef]
- Paaver, U.; Lust, A.; Mirza, S.; Rantanen, J.; Veski, P.; Heinamaki, J.; Kogermann, K. Insight into the solubility and dissolution behavior of piroxicam anhydrate and monohydrate forms. Int. J. Pharm. 2012, 431, 111–119. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paaver, U.; Heinämäki, J.; Kassamakov, I.; Ylitalo, T.; Hæggström, E.; Laidmäe, I.; Kogermann, K. Quasi-Dynamic Dissolution of Electrospun Polymeric Nanofibers Loaded with Piroxicam. Pharmaceutics 2019, 11, 491. https://doi.org/10.3390/pharmaceutics11100491
Paaver U, Heinämäki J, Kassamakov I, Ylitalo T, Hæggström E, Laidmäe I, Kogermann K. Quasi-Dynamic Dissolution of Electrospun Polymeric Nanofibers Loaded with Piroxicam. Pharmaceutics. 2019; 11(10):491. https://doi.org/10.3390/pharmaceutics11100491
Chicago/Turabian StylePaaver, Urve, Jyrki Heinämäki, Ivan Kassamakov, Tuomo Ylitalo, Edward Hæggström, Ivo Laidmäe, and Karin Kogermann. 2019. "Quasi-Dynamic Dissolution of Electrospun Polymeric Nanofibers Loaded with Piroxicam" Pharmaceutics 11, no. 10: 491. https://doi.org/10.3390/pharmaceutics11100491
APA StylePaaver, U., Heinämäki, J., Kassamakov, I., Ylitalo, T., Hæggström, E., Laidmäe, I., & Kogermann, K. (2019). Quasi-Dynamic Dissolution of Electrospun Polymeric Nanofibers Loaded with Piroxicam. Pharmaceutics, 11(10), 491. https://doi.org/10.3390/pharmaceutics11100491