Pharmacokinetics and Pharmacodynamics of (S)-Ketoprofen Co-Administered with Caffeine: A Preclinical Study in Arthritic Rats

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Animals
2.3. Pharmacokinetics of (S)-Ketoprofen
2.3.1. Blood Sampling
2.3.2. Sample Preparation
2.3.3. Chromatographic Conditions
2.4. Pharmacodynamics of (S)-Ketoprofen
2.4.1. Arthritis Induction
2.4.2. Behavioral Assessment
2.5. Pharmacokinetic and Pharmacodynamic Data Analysis
3. Results
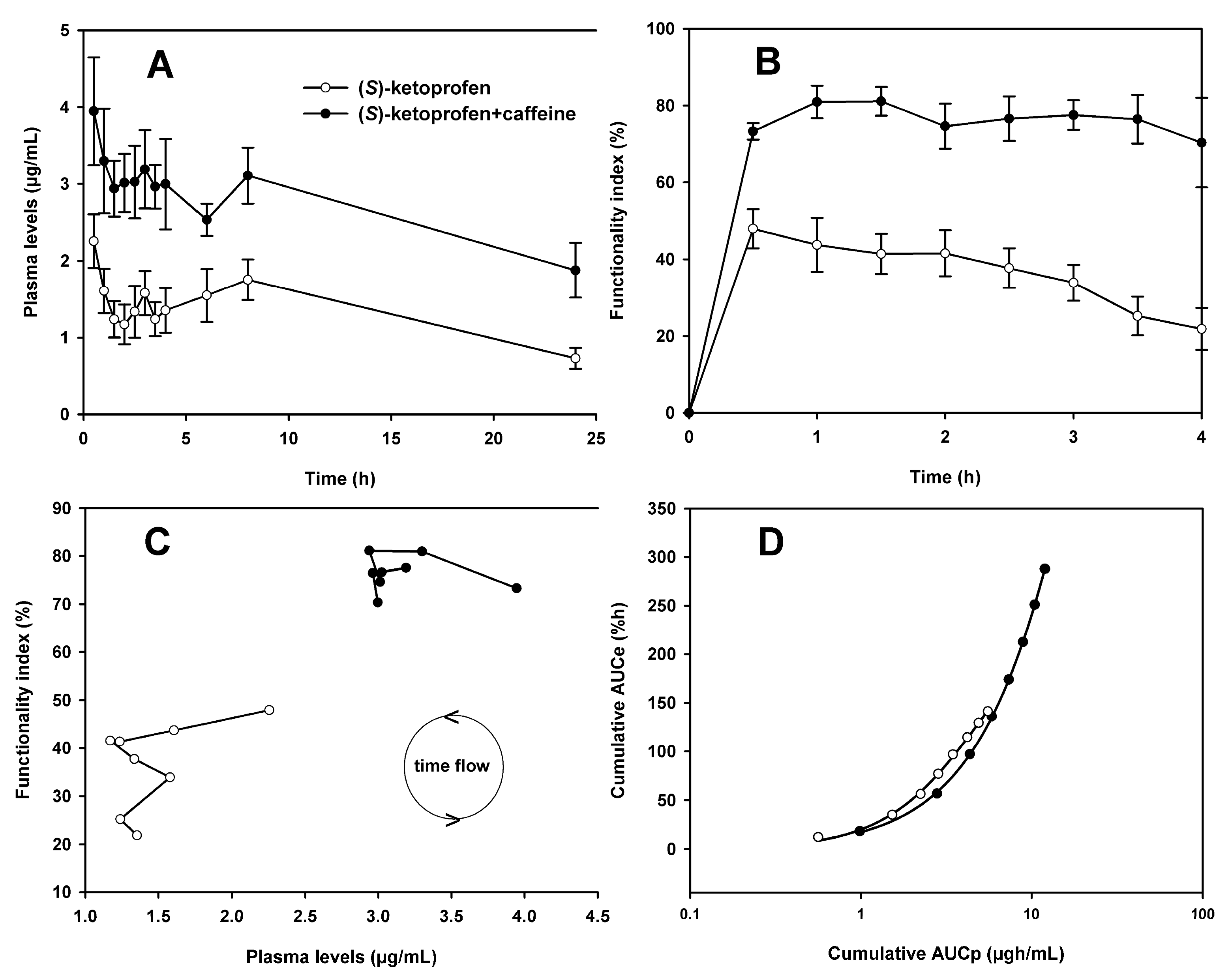
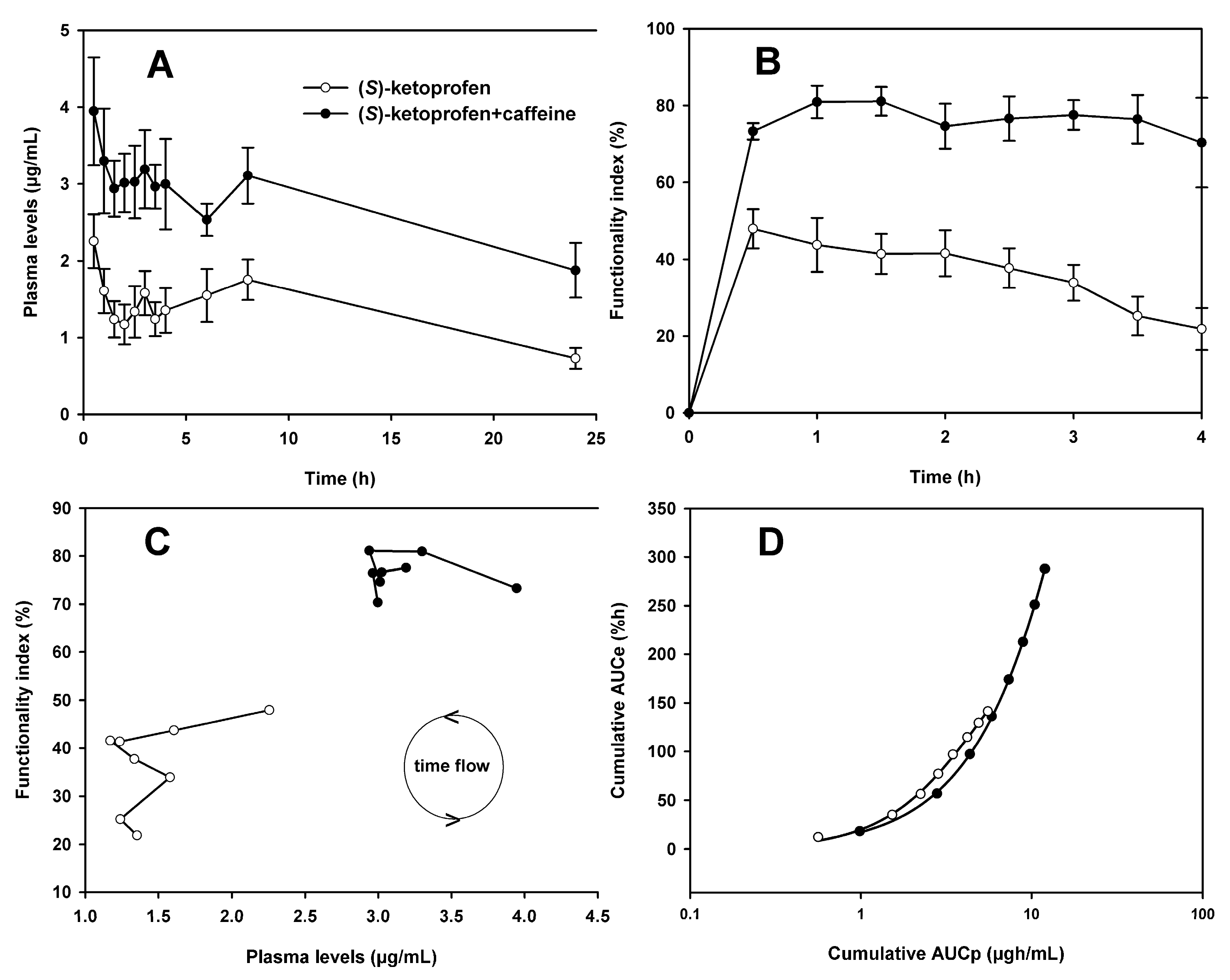
3.1. Pharmacokinetics of (S)-Ketoprofen
3.2. Pharmacodynamics of (S)-Ketoprofen
3.3. Pharmacokinetic and Pharmacodynamic Data Analysis
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Breimer, D.D. PK/PD modelling and beyond: Impact on drug development. Pharm. Res. 2008, 25, 2720–2722. [Google Scholar] [CrossRef] [PubMed]
- Rajman, I. PK/PD modelling and simulations: Utility in drug development. Drug Discov. Today 2008, 13, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Colbourn, W.A. Controversy III: To model or not to model. J. Clin. Pharmacol. 1988, 28, 879–888. [Google Scholar] [CrossRef]
- Lees, P.; Giraudel, J.; Landoni, M.F.; Toutain, P.L. PK-PD integration and PK-PD modelling of nonsteroidal anti-inflammatory drugs: Principles and applications in veterinary pharmacology. J. Vet. Pharmacol. Ther. 2004, 27, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Granados-Soto, V.; Flores-Murrieta, F.J.; Lopez-Munoz, F.J.; Salazar, L.A.; Villarreal, J.E.; Castaneda-Hernandez, G. Relationship between paracetamol plasma levels and its analgesic effect in the rat. J. Pharm. Pharmacol. 1992, 44, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Granados-Soto, V.; Lopez-Munoz, F.J.; Hong, E.; Flores-Murrieta, F.J. Relationship between pharmacokinetics and the analgesic effect of ketorolac in the rat. J. Pharmacol. Exp. Ther. 1995, 272, 352–356. [Google Scholar] [PubMed]
- Torres-Lopez, J.E.; Lopez-Munoz, F.J.; Castaneda-Hernandez, G.; Flores-Murrieta, F.J.; Granados-Soto, V. Pharmacokinetic-pharmacodynamic modeling of the antinociceptive effect of diclofenac in the rat. J. Pharmacol. Exp. Ther. 1997, 282, 685–690. [Google Scholar] [PubMed]
- Flores-Murrieta, F.J.; Ko, H.C.; Flores-Acevedo, D.M.; Lopez-Munoz, F.J.; Jusko, W.J.; Sale, M.E.; Castaneda-Hernandez, G. Pharmacokinetic-pharmacodynamic modeling of tolmetin antinociceptive effect in the rat using an indirect response model: A population approach. J. Pharmacokinet. Biopharm. 1998, 26, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Granados-Soto, V.; Lopez-Munoz, F.J.; Castaneda-Hernandez, G.; Salazar, L.A.; Villarreal, J.E.; Flores-Murrieta, F.J. Characterization of the analgesic effects of paracetamol and caffeine combinations in the pain-induced functional impairment model in the rat. J. Pharm. Pharmacol. 1993, 45, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Hernandez, G.; Castillo-Mendez, M.S.; Lopez-Munoz, F.J.; Granados-Soto, V.; Flores-Murrieta, F.J. Potentiation by caffeine of the analgesic effect of aspirin in the pain-induced functional impairment model in the rat. Can. J. Physiol. Pharmacol. 1994, 72, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Hoyo-Vadillo, C.; Perez-Urizar, J.; Lopez-Munoz, F.J. Usefulness of the pain-induced functional impairment model to relate plasma levels of analgesics to their efficacy in rats. J. Pharm. Pharmacol. 1995, 47, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Reval, M.I.; Ventura-Martinez, R.; Hernandez-Delgadillo, G.P.; Dominguez-Ramirez, A.M.; Lopez-Munoz, F.J. Effect of caffeine on antinociceptive action of ketoprofen in rats. Arch. Med. Res. 2001, 32, 13–20. [Google Scholar] [CrossRef]
- Lopez, J.R.M.; Dominguez-Ramirez, A.M.; Cook, H.J.; Bravo, G.; Diaz-Reval, M.I.; Deciga-Campos, M.; Lopez-Munoz, F.J. Enhancement of antinociception by co-administration of ibuprofen and caffeine in arthritic rats. Eur. J. Pharmacol. 2006, 544, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Munoz, F.J.; Ventura, R.; Diaz, M.I.; Fernandez-Guasti, A.; Tost, D.; Cabre, F.; Mauleon, D. Antinociceptive effects of S(+)-ketoprofen and other analgesic drugs in a rat model of pain induced by uric acid. J. Clin. Pharmacol. 1998, 38 (Suppl. 12), 11S–21S. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Toda, Y.; Hori, M.; Mitani, A.; Ichihara, T.; Sekine, S.; Kaku, S.; Otsuka, N.; Matsumoto, H. Topical anti-inflammatory and analgesic effects of multiple applications of S(+)-flurbiprofen plaster (SFPP) in a rat adjuvant-induced arthritis model. Drug Dev. Res. 2016, 77, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Komatsu, H.; Kawamoto, H.; Yamada, T. Studies on the effects of antiinflammatory action of benzoyl-hydrotropic acid (ketoprofen) and other drugs, with special reference to prostaglandin synthesis. Arch. Int. Pharmacodyn. Ther. 1979, 237, 169–176. [Google Scholar] [PubMed]
- Cabre, F.; Fernandez, M.F.; Calvo, L.; Ferrer, X.; Garcia, M.L.; Mauleon, D. Analgesic, antiinflammatory, and antipyretic effects of S(+)-ketoprofen in vivo. J. Clin. Pharmacol. 1998, 38 (Suppl. 12), 3S–10S. [Google Scholar] [CrossRef] [PubMed]
- Suesa, N.; Fernandez, M.F.; Gutierrez, M.; Rufat, M.J.; Rotllan, E.; Calvo, L.; Mauleon, D.; Carganico, G. Stereoselective cyclooxygenase inhibition in cellular models by the enantiomers of ketoprofen. Chirality 1993, 5, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Carabaza, A.; Cabre, F.; Rotllan, E.; Gomez, M.; Gutierrez, M.; Garcia, M.L.; Mauleon, D. Stereoselective inhibition of inducible cyclooxygenase by chiral nonsteroidal antiinflammatory drugs. J. Clin. Pharmacol. 1996, 36, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Miranda, H.F.; Puig, M.M.; Dursteler, C.; Prieto, J.C.; Pinardi, G. Dexketoprofen-induced antinociception in animal models of acute pain: Synergy with morphine and paracetamol. Neuropharmacology 2007, 52, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Morgaz, J.; Navarrete, R.; Munoz-Rascon, P.; Dominguez, J.M.; Fernandez-Sarmiento, J.A.; Gomez-Villamandos, R.J.; Granados, M.M. Postoperative analgesic effects of dexketoprofen, buprenorphine and tramadol in dogs undergoing ovariohysterectomy. Res. Vet. Sci. 2013, 95, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Pouchain, E.C.; Costa, F.W.G.; Bezerra, T.P.; Soares, E.C.S. Comparative efficacy of nimesulide and ketoprofen on inflammatory events in third molar surgery: A split-mouth, prospective, randomized, double-blind study. Int. J. Oral Maxillofac. Surg. 2015, 44, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Baratloo, A.; Forouzanfar, M.M.; Safari, S.; Rouhipour, A.; Amiri, M.; Negida, A. The role of caffeine in pain management: A brief literature review. Anesthesiol. Pain Med. 2016, 6, e33193. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Lieb, K.; Hull, M.; Aicher, B.; van Ryn, J.; Pairet, M.; Engelhardt, G. Effects of caffeine and paracetamol alone or in combination with acetylsalicylic acid on prostaglandin E2 synthesis in rat microglial cells. Neuropharmacology 2000, 39, 2205–2213. [Google Scholar] [CrossRef]
- Sawynok, J.; Reid, A.R. Caffeine inhibits antinociception by acetaminophen in the formalin test by inhibiting spinal adenosine A1 receptors. Eur. J. Pharmacol. 2012, 674, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Sawynok, J. Caffeine and pain. Pain 2011, 152, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ramos, J.M.; Diaz-Reval, M.I. Opioid mechanism involvement in the synergism produced by the combination of diclofenac and caffeine in the formalin model. ISRN Pain 2013, 2013, 196429. [Google Scholar] [CrossRef] [PubMed]
- Sawynok, J.; Yaksh, T.L. Caffeine as an analgesic adjuvant: A review of pharmacology and mechanisms of action. Pharmacol. Rev. 1993, 45, 43–85. [Google Scholar] [PubMed]
- Lopez-Munoz, F.J.; Salazar, L.A.; Castaneda-Hernandez, G.; Villarreal, J.E. A new model to assess analgesic activity: Pain-induced functional impairment in the rat (PIFIR). Drug Dev. Res. 1993, 28, 169–175. [Google Scholar] [CrossRef]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Lopez-Munoz, F.J.; Gama, N.V.; Soria-Arteche, O.; de la Pena, M.H.Y.; Dominguez-Ramirez, A.M.; Lopez, J.R.M. HPLC method with solid-phase extraction for determination of (R)- and (S)-ketoprofen in plasma without caffeine interference: Application to pharmacokinetic studies in rats. J. Chromatogr. Sci. 2014, 52, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics and Pharmacodinamics: Concepts and Applications, 2nd ed.; Lea and Febiger: Philadelphia, PA, USA, 1988; pp. 459–463. ISBN 0812111605. [Google Scholar]
- Dominguez-Ramirez, A.M.; Cortes-Arroyo, A.R.; Y de la Pena, M.H.; Lopez, J.R.M.; Lopez-Munoz, F.J. Effect of metamizol on morphine pharmacokinetics and pharmacodynamics after acute and subchronic administration in arthritic rats. Eur. J. Pharmacol. 2010, 645, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, K.; Niemi, A.; Raekallio, M.; Heinonen, M.; Peltoniemi, O.A.T.; Palviainen, M.; Siven, M.; Peltoniemi, M.; Vainio, O. Enantiospecific ketoprofen concentrations in plasma after oral and intramuscular administration in growing pigs. Acta Vet. Scand. 2012, 54, 55. [Google Scholar] [CrossRef] [PubMed]
- Yasui, H.; Yamaoka, K.; Nakagawa, T. Moment analysis of stereoselective enterohepatic circulation and unidirectional chiral inversion of ketoprofen enantiomers in rat. J. Pharm. Sci. 1996, 85, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Raekallio, M.R.; Mustonen, K.M.; Heinonen, M.L.; Peltoniemi, O.A.T.; Sakkinen, M.S.; Peltoniemi, S.M.; Honkavaara, J.M.; Vainio, O.M. Evaluation of bioequivalence after oral, intramuscular, and intravenous administration of racemic ketoprofen in pigs. Am. J. Vet. Res. 2008, 69, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Jamali, F.; Brocks, D.R. Clinical pharmacokinetics of ketoprofen and its enantiomers. Clin. Pharmacokinet. 1990, 19, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, S.; He, X.; Hashimoto, S.; Volland, C.; Benet, L.Z.; Lin, E.T. Stereoselective disposition of ketoprofen in rats. Drug Metab. Dispos. 1991, 19, 717–718. [Google Scholar] [PubMed]
- Daly, J.W. Mechanism of Action of Caffeine; Raven: Norris, MT, USA, 1993; pp. 97–150. [Google Scholar]
- Biaggioni, I.; Paul, S.; Puckett, A.; Arzubiaga, C. Caffeine and theophylline as adenosine receptor antagonists in humans. J. Pharmacol. Exp. Ther. 1991, 258, 588–593. [Google Scholar] [PubMed]
- López-Muñoz, F.J.; Vara-Gama, N.; Soria-Arteche, O.; Domínguez-Ramírez, A.M.; Medina, R. Enhancement of (S)-ketoprofen antinociception by caffeine co-administration: Effects in an arthritic-pain animal model. Lat. Am. J. Pharm. 2017, 36, 1349–1354. [Google Scholar]
- Lim, R.K. Pain. Annu. Rev. Physiol. 1970, 32, 269–288. [Google Scholar] [CrossRef] [PubMed]
- De Beaurepaire, R.; Suaudeau, C.; Chait, A.; Cimetiere, C. Anatomical mapping of brain sites involved in the antinociceptive effects of ketoprofen. Brain Res. 1990, 536, 201–206. [Google Scholar] [CrossRef]
- Braga, P.C. Ketoprofen: i.c.v. Injection and electrophysiological aspects of antinociceptive effect. Eur. J. Pharmacol. 1990, 184, 273–280. [Google Scholar] [CrossRef]
- Netter, P.; Lapicque, F.; Bannwarth, B.; Tamisier, J.N.; Thomas, P.; Royer, R.J. Diffusion of intramuscular ketoprofen into the cerebrospinal fluid. Eur. J. Clin. Pharmacol. 1985, 29, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Louizos, C.; Yanez, J.A.; Forrest, M.L.; Davies, N.M. Understanding the hysteresis loop conundrum in pharmacokinetic/pharmacodynamic relationships. J. Pharm. Pharm. Sci. 2014, 17, 34–91. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.M.; Liu, M.; Davis, I.M.; Shaw, S.; Yin, Y. Pharmacokinetics and pharmacodynamics of ranitidine in renal impairment. Eur. J. Clin. Pharmacol. 1997, 52, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Siegers, C.P. Effects of caffeine on the absorption and analgesic efficacy of paracetamol in rats. Pharmacology 1973, 10, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Flores-Acevedo, D.M.; Flores-Murrieta, F.J.; Castaneda-Hernandez, G.; Lopez-Munoz, F.J. Potentiation of the analgesic effect of tolmetin, a potent non-steroid anti-inflammatory drug, by caffeine in the rat. Pharm. Sci. 1995, 1, 441–444. [Google Scholar] [CrossRef]

{kind=link}
| Pharmacokinetics | (S)-Ketoprofen | (S)-Ketoprofen + Caffeine |
|---|---|---|
| Cmax (μg/mL) | 2.73 ± 0.27 | 5.19 ± 0.59 * |
| Tmax (h) | 0.50 ± 0.00 | 0.50 ± 0.00 |
| AUC0-24 (μg·h/mL) | 34.49 ± 6.11 | 63.03 ± 7.66 * |
| AUC0-∞ (μg·h/mL) | 71.17 ± 18.58 | 112.17 ± 24.78 * |
| λz (h−1) | 0.0389 ± 0.01 | 0.0404 ± 0.01 |
| t½ (h) | 26.94 ± 9.22 | 18.45 ± 3.45 |
| MRT (h) | 38.38 ± 12.72 | 26.66 ± 5.71 |
| Pharmacodynamics | ||
| Emax (%) | 47.93 ± 5.07 | 81.09 ± 3.74 * |
| Tmax (h) | 0.83 ± 0.25 | 2.0 ± 0.52 * |
| AUC0-4 (%h) | 141.35 ± 18.09 | 287.80 ± 14.02 * |
| Sigmoidal Emax model | ||
| Emax (%h) | 252.21 ± 27.27 | 1486.97 ± 463.58 * |
| EC50 (μg·h/mL) | 5.49 ± 1.58 | 43.67 ± 21.45 * |
| Response factor (γ) | 1.64 ± 0.16 | 1.25 ± 0.10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-López, R.; Vara-Gama, N.; Soria-Arteche, O.; Moreno-Rocha, L.A.; López-Muñoz, F.J. Pharmacokinetics and Pharmacodynamics of (S)-Ketoprofen Co-Administered with Caffeine: A Preclinical Study in Arthritic Rats. Pharmaceutics 2018, 10, 20. https://doi.org/10.3390/pharmaceutics10010020
Medina-López R, Vara-Gama N, Soria-Arteche O, Moreno-Rocha LA, López-Muñoz FJ. Pharmacokinetics and Pharmacodynamics of (S)-Ketoprofen Co-Administered with Caffeine: A Preclinical Study in Arthritic Rats. Pharmaceutics. 2018; 10(1):20. https://doi.org/10.3390/pharmaceutics10010020
Chicago/Turabian StyleMedina-López, Raúl, Nancy Vara-Gama, Olivia Soria-Arteche, Luis A. Moreno-Rocha, and Francisco J. López-Muñoz. 2018. "Pharmacokinetics and Pharmacodynamics of (S)-Ketoprofen Co-Administered with Caffeine: A Preclinical Study in Arthritic Rats" Pharmaceutics 10, no. 1: 20. https://doi.org/10.3390/pharmaceutics10010020
APA StyleMedina-López, R., Vara-Gama, N., Soria-Arteche, O., Moreno-Rocha, L. A., & López-Muñoz, F. J. (2018). Pharmacokinetics and Pharmacodynamics of (S)-Ketoprofen Co-Administered with Caffeine: A Preclinical Study in Arthritic Rats. Pharmaceutics, 10(1), 20. https://doi.org/10.3390/pharmaceutics10010020