
Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Multiple Pathways to Death
2.1. Apoptosis
2.2. Necroptosis
2.3. Pyroptosis
2.4. Cross-Talk between Death Pathways
3. Activation of Cell Death Pathways by CMV
3.1. Direct Recognition of CMV Infection
3.2. Cellular Stress Responses
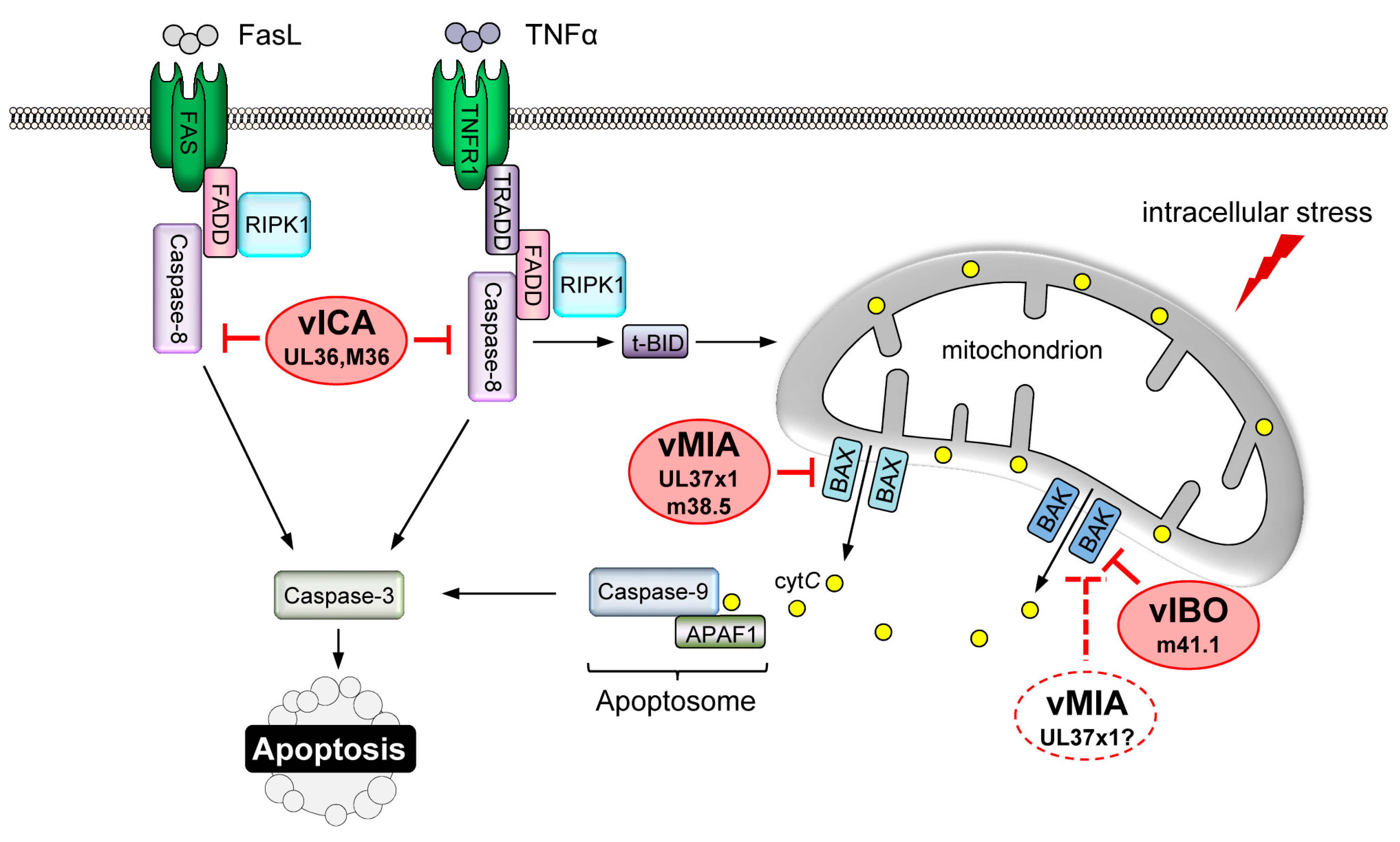
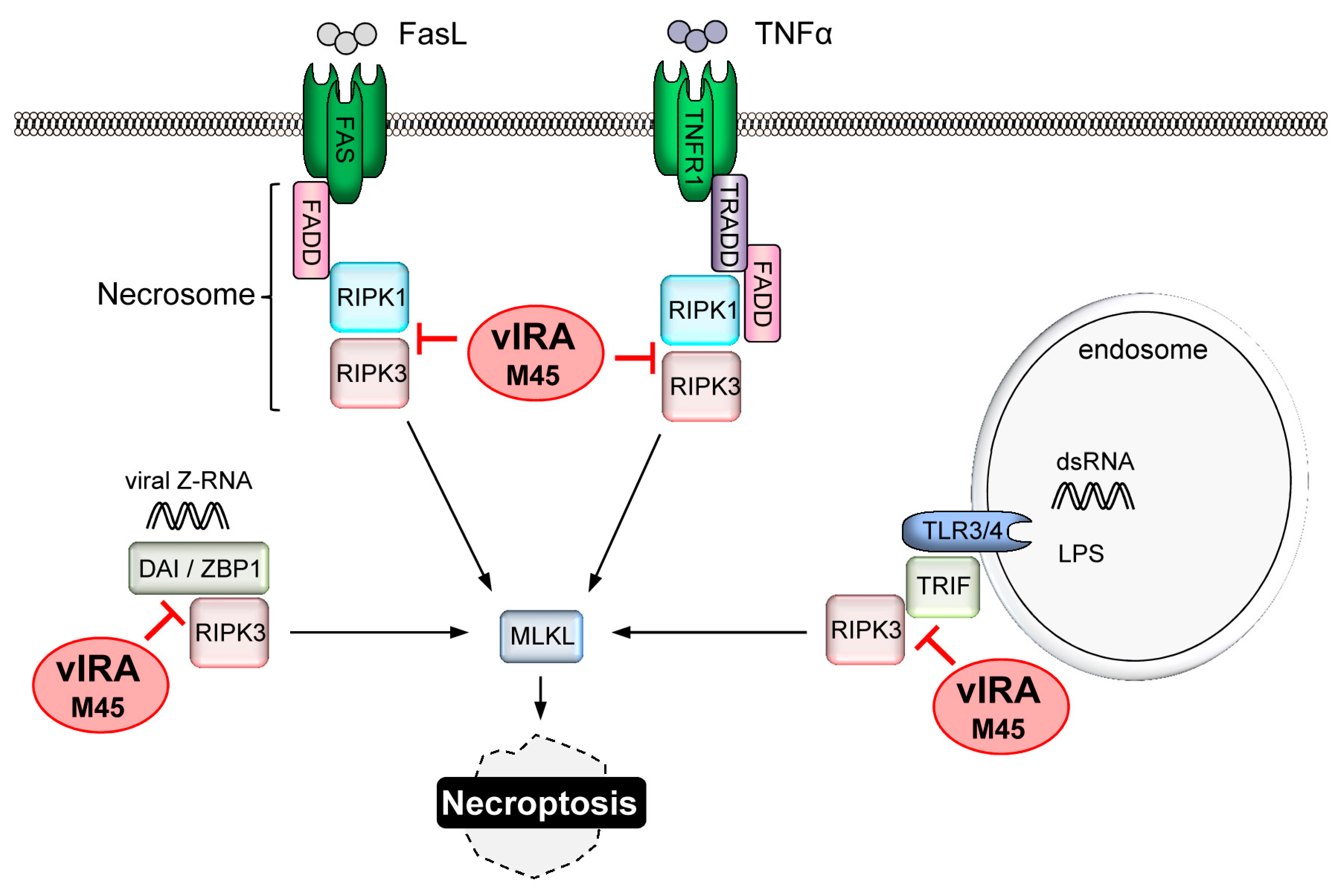
4. CMV-Encoded Death Inhibitors
4.1. Inhibition of MOMP
4.2. Suppression of the ER Stress Response
4.3. Inhibition of DR-Mediated Apoptosis
4.4. Replication of CMV Requires Suppression of Necroptosis
4.5. Interference with Pyroptosis?
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Dillon, C.P.; Green, D.R. Molecular Cell Biology of Apoptosis and Necroptosis in Cancer. Adv. Exp. Med. Biol. 2016, 930, 1–23. [Google Scholar] [PubMed]
- Nagata, S.; Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Tuzlak, S.; Kaufmann, T.; Villunger, A. Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis. Genes Dev. 2016, 30, 2133–2151. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, H.; Upton, J.W. Programmed necrosis in microbial pathogenesis. Trends Microbiol. 2014, 22, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1960–2014. [Google Scholar]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Meusburger, S.; Hawkins, C.J.; Riglar, D.T.; Lee, E.F.; Fairlie, W.D.; Huang, D.C.; Adams, J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. USA 2008, 105, 18081–18087. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Westphal, D.; Dewson, G.; Ma, S.; Hockings, C.; Fairlie, W.D.; Lee, E.F.; Yao, S.; Robin, A.Y.; Smith, B.J.; et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 2013, 152, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Grace, C.R.; Llambi, F.; Nourse, A.; Fitzgerald, P.; Gehring, K.; Kriwacki, R.W.; Green, D.R. BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 2013, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Lindsten, T.; Zong, W.X.; Cinalli, R.M.; Thompson, C.B. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat. Immunol. 2002, 3, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Boatright, K.M.; Renatus, M.; Scott, F.L.; Sperandio, S.; Shin, H.; Pedersen, I.M.; Ricci, J.E.; Edris, W.A.; Sutherlin, D.P.; Green, D.R.; et al. A unified model for apical caspase activation. Mol. Cell 2003, 11, 529–541. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-α-induced apoptosis by NF-κB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Oberst, A.; Drag, M.; van Raam, B.J.; Riedl, S.J.; Green, D.R.; Salvesen, G.S. FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 2011, 433, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, X.; McQuade, T.; Li, J.; Chan, F.K.; Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Weinlich, R.; Rodriguez, D.A.; Cripps, J.G.; Quarato, G.; Gurung, P.; Verbist, K.C.; Brewer, T.L.; Llambi, F.; Gong, Y.N.; et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Rickard, J.A.; O’Donnell, J.A.; Evans, J.M.; Lalaoui, N.; Poh, A.R.; Rogers, T.; Vince, J.E.; Lawlor, K.E.; Ninnis, R.L.; Anderton, H.; et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 2014, 157, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Daley-Bauer, L.P.; Thapa, R.J.; Mandal, P.; Berger, S.B.; Huang, C.; Sundararajan, A.; Guo, H.; Roback, L.; Speck, S.H.; et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. USA 2014, 111, 7753–7758. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed]
- Szomolanyi-Tsuda, E.; Liang, X.; Welsh, R.M.; Kurt-Jones, E.A.; Finberg, R.W. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J. Virol. 2006, 80, 4286–4291. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3516–3521. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Estornes, Y.; Toscano, F.; Virard, F.; Jacquemin, G.; Pierrot, A.; Vanbervliet, B.; Bonnin, M.; Lalaoui, N.; Mercier-Gouy, P.; Pacheco, Y.; et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012, 19, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Liverpool, L.; Bridgeman, A.; Ragan, K.B.; Upton, J.W.; Rehwinkel, J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, H.; Ragan, K.B.; Guo, H.; Gilley, R.P.; Landsteiner, V.J.; Kaiser, W.J.; Upton, J.W. Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep. 2017, 18, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Valchanova, R.S.; Picard-Maureau, M.; Budt, M.; Brune, W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J. Virol. 2006, 80, 10181–10190. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Wolff, T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses 2009, 1, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Alcami, J.; Esteban, M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the α subunit of eukaryotic translation initiation factor 2 and NF-κB. Mol. Cell Biol. 1999, 19, 4653–4663. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Kim, C.N.; Yeh, W.C.; Mak, T.W.; Bhalla, K.; Barber, G.N. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998, 17, 6888–6902. [Google Scholar] [CrossRef] [PubMed]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine cytomegalovirus targets transcription factor ATF4 to exploit the unfolded-protein response. J. Virol. 2012, 86, 6712–6723. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Xiaofei, E.; Kowalik, T.F. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 2014, 6, 2155–2185. [Google Scholar] [PubMed]
- Gaspar, M.; Shenk, T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 2821–2826. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Rosenke, K.; Czornak, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.; Han, J.; Lutz, R.J.; Watanabe, S.; McFarland, E.D.; et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 1999, 96, 12536–12541. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.; Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA 2004, 101, 7988–7993. [Google Scholar] [CrossRef] [PubMed]
- Poncet, D.; Larochette, N.; Pauleau, A.L.; Boya, P.; Jalil, A.A.; Cartron, P.F.; Vallette, F.; Schnebelen, C.; Bartle, L.M.; Skaletskaya, A.; et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J. Biol. Chem. 2004, 279, 22605–22614. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.L.; Youle, R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 2008, 82, 6232–6243. [Google Scholar] [CrossRef] [PubMed]
- Sharon-Friling, R.; Goodhouse, J.; Colberg-Poley, A.M.; Shenk, T. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc. Natl. Acad. Sci. USA 2006, 103, 19117–19122. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Ferreira, A.R.; Gomes, S.; Vieira, M.; Gouveia, A.; Valenca, I.; Islinger, M.; Nascimento, R.; Schrader, M.; Kagan, J.C.; et al. Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep. 2016, 6, 26028. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Schumacher, U.; Simic, H.; Voigt, S.; Brune, W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J. Virol. 2008, 82, 4812–4822. [Google Scholar] [CrossRef] [PubMed]
- Manzur, M.; Fleming, P.; Huang, D.C.; Degli-Esposti, M.A.; Andoniou, C.E. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 2009, 16, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Skaletskaya, A.; Estaquier, J.; Dufour, C.; Goldmacher, V.S. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis 2008, 13, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Çam, M.; Handke, W.; Picard-Maureau, M.; Brune, W. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 2010, 17, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.; Kvansakul, M.; Voigt, V.; Kile, B.T.; Kluck, R.M.; Huang, D.C.; Degli-Esposti, M.A.; Andoniou, C.E. MCMV-mediated inhibition of the pro-apoptotic Bak protein is required for optimal in vivo replication. PLoS Pathog. 2013, 9, e1003192. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.N.; McCormick, A.L.; Mocarski, E.S. Gene products of the embedded m41/m41.1 locus of murine cytomegalovirus differentially influence replication and pathogenesis. Virology 2013, 436, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Handke, W.; Luig, C.; Popovic, B.; Krmpotic, A.; Jonjic, S.; Brune, W. Viral inhibition of BAK promotes murine cytomegalovirus dissemination to salivary glands. J. Virol. 2013, 87, 3592–3596. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Cao, X. GRIM-19 is essential for maintenance of mitochondrial membrane potential. Mol. Biol. Cell 2008, 19, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J. Virol. 2011, 85, 9103–9113. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.; Burkhart, J.M.; Hinte, F.; Tirosh, B.; Mohr, H.; Zahedi, R.P.; Sickmann, A.; Ruzsics, Z.; Budt, M.; Brune, W. Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 2013, 9, e1003544. [Google Scholar] [CrossRef] [PubMed]
- Seirafian, S.; Prod’homme, V.; Sugrue, D.; Davies, J.; Fielding, C.; Tomasec, P.; Wilkinson, G.W. Human cytomegalovirus suppresses Fas expression and function. J. Gen. Virol. 2014, 95, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Popkin, D.L.; Virgin, H.W., 4th. Murine cytomegalovirus infection inhibits tumor necrosis factor α responses in primary macrophages. J. Virol. 2003, 77, 10125–10130. [Google Scholar] [CrossRef] [PubMed]
- Le, V.T.; Trilling, M.; Hengel, H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb'-encoded modulation of TNF-α signaling. J. Virol. 2011, 85, 13260–13270. [Google Scholar] [CrossRef] [PubMed]
- Montag, C.; Wagner, J.A.; Gruska, I.; Vetter, B.; Wiebusch, L.; Hagemeier, C. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor α (TNF-α) signaling by upregulating TNF-α receptor 1 cell surface expression. J. Virol. 2011, 85, 11409–11421. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Loewendorf, A.; Wang, Q.; McDonald, B.; Redwood, A.; Benedict, C.A. Inhibition of the TRAIL death receptor by CMV reveals its importance in NK cell-mediated antiviral defense. PLoS Pathog. 2014, 10, e1004268. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Tomasec, P.; Aicheler, R.; Loewendorf, A.; Nemcovicova, I.; Wang, E.C.; Stanton, R.J.; Macauley, M.; Norris, P.; Willen, L.; et al. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 2013, 13, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.L.; Skaletskaya, A.; Barry, P.A.; Mocarski, E.S.; Goldmacher, V.S. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 2003, 316, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Wagner, M.; Ruzsics, Z.; Holak, K.; Brune, W.; Campbell, A.E.; Koszinowski, U.H. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 2003, 77, 5557–5570. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.; Kasmapour, B.; Plaza, C.; Bajagic, M.; Casalegno Garduno, R.; Borkner, L.; Lenac Roviš, T.; Scrima, A.; Jonjic, S.; Schmitz, I.; et al. UL36 Rescues Apoptosis Inhibition and In Vivo Replication of A Chimeric MCMV Lacking the M36 Gene. Front. Microbiol. 2017, 7, 312. [Google Scholar] [CrossRef] [PubMed]
- Cicin-Sain, L.; Ruzsics, Z.; Podlech, J.; Bubic, I.; Menard, C.; Jonjic, S.; Reddehase, M.J.; Koszinowski, U.H. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J. Virol. 2008, 82, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Hüttmann, J.; Krause, E.; Schommartz, T.; Brune, W. Functional Comparison of Molluscum Contagiosum Virus vFLIP MC159 with Murine Cytomegalovirus M36/vICA and M45/vIRA Proteins. J. Virol. 2015, 90, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Ebermann, L.; Ruzsics, Z.; Guzman, C.A.; van Rooijen, N.; Casalegno-Garduno, R.; Koszinowski, U.; Cicin-Sain, L. Block of death-receptor apoptosis protects mouse cytomegalovirus from macrophages and is a determinant of virulence in immunodeficient hosts. PLoS Pathog. 2012, 8, e1003062. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.L.; Roback, L.; Livingston-Rosanoff, D.; St Clair, C. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J. Virol. 2010, 84, 5108–5123. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Ménard, C.; Heesemann, J.; Koszinowski, U.H. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 2001, 291, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Lembo, D.; Donalisio, M.; Hofer, A.; Cornaglia, M.; Brune, W.; Koszinowski, U.; Thelander, L.; Landolfo, S. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J. Virol. 2004, 78, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Krause, E.; de Graaf, M.; Fliss, P.M.; Dölken, L.; Brune, W. Murine cytomegalovirus virion-associated protein M45 mediates rapid NF-κB activation after infection. J. Virol. 2014, 88, 9963–9975. [Google Scholar] [CrossRef] [PubMed]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef] [PubMed]
- Daley-Bauer, L.P.; Roback, L.; Crosby, L.N.; McCormick, A.L.; Feng, Y.; Kaiser, W.J.; Mocarski, E.S. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E2786–E2795. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Guo, H.; Talekar, G.R.; Roback, L.; Kaiser, W.J.; Mocarski, E.S. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J. Biol. Chem. 2015, 290, 11635–11648. [Google Scholar] [CrossRef] [PubMed]
- Lembo, D.; Brune, W. Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 2009, 34, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, S.Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, D.; Huang, H.; Lu, Y.; Liao, Y.; Liu, L.; Liu, X.; Fang, F. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Sester, D.P.; Sagulenko, V.; Thygesen, S.J.; Cridland, J.A.; Loi, Y.S.; Cridland, S.O.; Masters, S.L.; Genske, U.; Hornung, V.; Andoniou, C.E.; et al. Deficient NLRP3 and AIM2 Inflammasome Function in Autoimmune NZB Mice. J. Immunol. 2015, 195, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Brune, W. Induction of apoptosis limits cytomegalovirus cross-species infection. EMBO J. 2006, 25, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; McGregor Dallas, S.R.; Colston, J.; Joseph, R.S.; Sinclair, J. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34+ progenitors. J. Gen. Virol. 2011, 92, 1539–1549. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. https://doi.org/10.3390/v9090249
Brune W, Andoniou CE. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses. 2017; 9(9):249. https://doi.org/10.3390/v9090249
Chicago/Turabian StyleBrune, Wolfram, and Christopher E. Andoniou. 2017. "Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus" Viruses 9, no. 9: 249. https://doi.org/10.3390/v9090249
APA StyleBrune, W., & Andoniou, C. E. (2017). Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses, 9(9), 249. https://doi.org/10.3390/v9090249