
Epigenetic Alterations in Human Papillomavirus-Associated Cancers


{kind=link}
Abstract
:1. Introduction
Human Papillomaviruses
2. DNA Methylation
2.1. HPV Genome Methylation
2.2. Cellular Gene Methylation
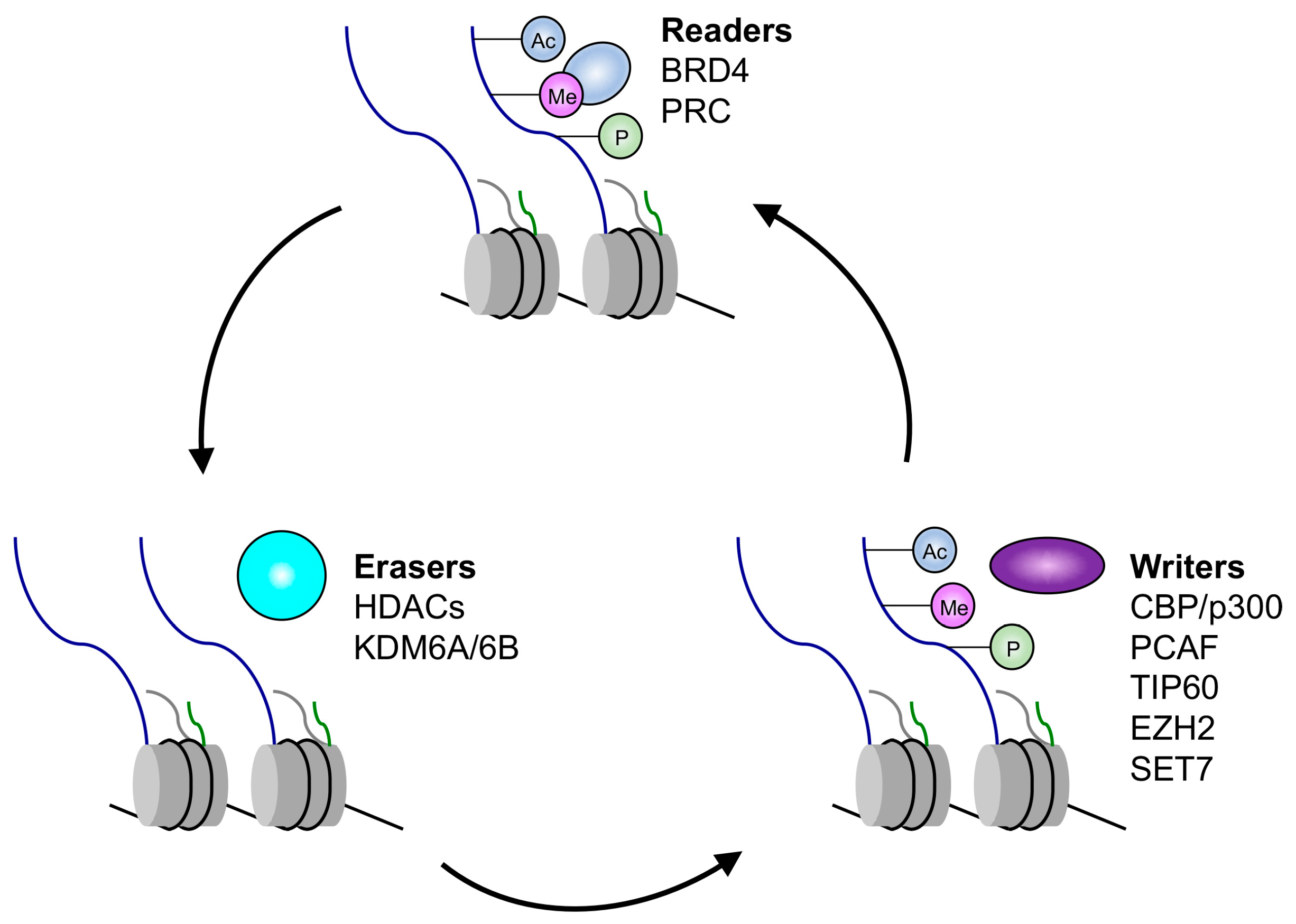
3. Regulation of Histone Modifications
3.1. Histone Modification of the Human Papillomavirus Genome
3.2. Acetylation
3.3. Polycomb Group Proteins and Histone Lysine Modifications
3.4. Histone Arginine Modifications
3.5. Epigenetic Readers
4. Non-Coding RNAs
4.1. MicroRNAs
4.2. Long Non-Coding RNAs
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses associated with human cancer. Biochim. Biophys. Acta 2008, 1782, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [PubMed]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef] [PubMed]
- Szalmas, A.; Konya, J. Epigenetic alterations in cervical carcinogenesis. Semin. Cancer Biol. 2009, 19, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Poreba, E.; Broniarczyk, J.K.; Gozdzicka-Jozefiak, A. Epigenetic mechanisms in virus-induced tumorigenesis. Clin. Epigenet. 2011, 2, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Minarovits, J.; Niller, H. Patho-epigenetics. Med. Epigenet. 2013, 1, 37–45. [Google Scholar] [CrossRef]
- Fernandez, A.F.; Esteller, M. Viral epigenomes in human tumorigenesis. Oncogene 2010, 29, 1405–1420. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodríguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Gillison, M.L.; Lowy, D.R. A causal role for human papillomavirus in head and neck cancer. Lancet 2004, 363, 1488–1489. [Google Scholar] [CrossRef]
- Frazer, I.H. Prevention of cervical cancer through papillomavirus vaccination. Nat. Rev. Immunol. 2004, 4, 46–54. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J. Aberrant DNA methylation in cervical carcinogenesis. Chin. J. Cancer 2013, 32, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Davis, J.N.; Day, M.L. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res. 2005, 65, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.L.; Tsang, T.Y.; Yau, P.L.; Kwok, T.T. Human papillomavirus type 16 E6 suppresses microRNA-23b expression in human cervical cancer cells through DNA methylation of the host gene C9orf3. Oncotarget 2017, 8, 12158–12173. [Google Scholar] [PubMed]
- Laurson, J.; Khan, S.; Chung, R.; Cross, K.; Raj, K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 2010, 31, 918–926. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef] [PubMed]
- Sartor, M.A.; Dolinoy, D.C.; Jones, T.R.; Colacino, J.A.; Prince, M.E.; Carey, T.E.; Rozek, L.S. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 2011, 6, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.M.; Wei, W.; Collins, S.I.; Pereira, M.; Diyaf, A.; Constandinou-Williams, C.; Young, L.S.; Roberts, S.; Woodman, C.B. Oncogenic human papillomavirus imposes an instructive pattern of DNA methylation changes which parallel the natural history of cervical HPV infection in young women. Carcinogenesis 2012, 33, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Doerfler, W.; Remus, R.; Muller, K.; Heller, H.; Hohlweg, U.; Schubbert, R. The fate of foreign DNA in mammalian cells and organisms. Dev. Biol. 2001, 106, 89–97. [Google Scholar]
- Fernandez, A.F.; Rosales, C.; Lopez-Nieva, P.; Grana, O.; Ballestar, E.; Ropero, S.; Espada, J.; Melo, S.A.; Lujambio, A.; Fraga, M.F.; et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009, 19, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.C.; Chiang, M.H.; Lai, H.C.; Hsiung, C.A.; Hsieh, C.Y.; Chu, T.Y. Methylation of the long control region of HPV16 is related to the severity of cervical neoplasia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 147, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Ye, F.; Lu, W.; Hu, Y.; Wan, X.; Chen, Y.; Xie, X. Methylation status of the long control region of HPV 16 in clinical cervical specimens. Mol. Med. Rep. 2008, 1, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Garner-Hamrick, P.A.; Fisher, C.; Lee, D.; Lambert, P.F. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J. Virol. 2003, 77, 12450–12459. [Google Scholar] [CrossRef] [PubMed]
- Badal, S.; Badal, V.; Calleja-Macias, I.E.; Kalantari, M.; Chuang, L.S.; Li, B.F.; Bernard, H.U. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology 2004, 324, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, M.; Calleja-Macias, I.E.; Tewari, D.; Hagmar, B.; Lie, K.; Barrera-Saldana, H.A.; Wiley, D.J.; Bernard, H.U. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J. Virol. 2004, 78, 12762–12772. [Google Scholar] [CrossRef] [PubMed]
- Turan, T.; Kalantari, M.; Calleja-Macias, I.E.; Cubie, H.A.; Cuschieri, K.; Villa, L.L.; Skomedal, H.; Barrera-Saldana, H.A.; Bernard, H.U. Methylation of the human papillomavirus-18 l1 gene: A biomarker of neoplastic progression? Virology 2006, 349, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, E.; Lambert, P.F. Epigenetics of human papillomaviruses. Virology 2013, 445, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, S.; von Knebel Doeberitz, M. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS ONE 2011, 6, e24451. [Google Scholar] [CrossRef] [PubMed]
- Mazumder Indra, D.; Singh, R.K.; Mitra, S.; Dutta, S.; Chakraborty, C.; Basu, P.S.; Mondal, R.K.; Roychoudhury, S.; Panda, C.K. Genetic and epigenetic changes of HPV16 in cervical cancer differentially regulate E6/E7 expression and associate with disease progression. Gynecol. Oncol. 2011, 123, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Hublarova, P.; Hrstka, R.; Rotterova, P.; Rotter, L.; Coupkova, M.; Badal, V.; Nenutil, R.; Vojtesek, B. Prediction of human papillomavirus 16 E6 gene expression and cervical intraepithelial neoplasia progression by methylation status. Int. J. Gynecol. Cancer 2009, 19, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Badal, V.; Chuang, L.S.; Tan, E.H.; Badal, S.; Villa, L.L.; Wheeler, C.M.; Li, B.F.; Bernard, H.U. Cpg methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: Genomic hypomethylation correlates with carcinogenic progression. J. Virol. 2003, 77, 6227–6234. [Google Scholar] [CrossRef] [PubMed]
- Snellenberg, S.; Schutze, D.M.; Claassen-Kramer, D.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.D. Methylation status of the E2 binding sites of HPV16 in cervical lesions determined with the Luminex xMAP system. Virology 2012, 422, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, M.; Lee, D.; Calleja-Macias, I.E.; Lambert, P.F.; Bernard, H.U. Effects of cellular differentiation, chromosomal integration and 5-aza-2′-deoxycytidine treatment on human papillomavirus-16 DNA methylation in cultured cell lines. Virology 2008, 374, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Chaiwongkot, A.; Vinokurova, S.; Pientong, C.; Ekalaksananan, T.; Kongyingyoes, B.; Kleebkaow, P.; Chumworathayi, B.; Patarapadungkit, N.; Reuschenbach, M.; von Knebel Doeberitz, M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int. J. Cancer 2013, 132, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Thain, A.; Jenkins, O.; Clarke, A.R.; Gaston, K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J. Virol. 1996, 70, 7233–7235. [Google Scholar] [PubMed]
- Marongiu, L.; Godi, A.; Parry, J.V.; Beddows, S. Human papillomavirus 16, 18, 31 and 45 viral load, integration and methylation status stratified by cervical disease stage. BMC Cancer 2014, 14, 384. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Gonzalez, A.; Lizano, M.; Candelaria, M.; Cetina, L.; Arce, C.; Cervera, E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol. Cancer 2005, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Sherman, M.E.; Schiffman, M.; Wang, S.S. Utility of methylation markers in cervical cancer early detection: Appraisal of the state-of-the-science. Gynecol. Oncol. 2009, 112, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Brebi, P.; Maldonado, L.; Noordhuis, M.G.; Ili, C.; Leal, P.; Garcia, P.; Brait, M.; Ribas, J.; Michailidi, C.; Perez, J.; et al. Genome-wide methylation profiling reveals Zinc finger protein 516 (ZNF516) and FK-506-binding protein 6 (FKBP6) promoters frequently methylated in cervical neoplasia, associated with HPV status and ethnicity in a chilean population. Epigenetics 2014, 9, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.L.; Chang, C.C.; Su, P.H.; Chen, Y.C.; Liao, Y.P.; Wang, H.C.; Yo, Y.T.; Chao, T.K.; Huang, H.C.; Lin, C.Y.; et al. Methylomic analysis identifies frequent DNA methylation of zinc finger protein 582 (ZNF582) in cervical neoplasms. PLoS ONE 2012, 7, e41060. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, K.P.; Brebi, P.M.; Roa, J.C. Epigenetic alterations in preneoplastic and neoplastic lesions of the cervix. Clin. Epigenet. 2012, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Hansel, A.; Steinbach, D.; Greinke, C.; Schmitz, M.; Eiselt, J.; Scheungraber, C.; Gajda, M.; Hoyer, H.; Runnebaum, I.B.; Durst, M. A promising DNA methylation signature for the triage of high-risk human papillomavirus DNA-positive women. PLoS ONE 2014, 9, e91905. [Google Scholar] [CrossRef] [PubMed]
- Siegel, E.M.; Riggs, B.M.; Delmas, A.L.; Koch, A.; Hakam, A.; Brown, K.D. Quantitative DNA methylation analysis of candidate genes in cervical cancer. PLoS ONE 2015, 10, e0122495. [Google Scholar] [CrossRef] [PubMed]
- Kitkumthorn, N.; Yanatatsanajit, P.; Kiatpongsan, S.; Phokaew, C.; Triratanachat, S.; Trivijitsilp, P.; Termrungruanglert, W.; Tresukosol, D.; Niruthisard, S.; Mutirangura, A. Cyclin A1 promoter hypermethylation in human papillomavirus-associated cervical cancer. BMC Cancer 2006, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, J.; Kooter, J.M.; Overmeer, R.M.; Claassen-Kramer, D.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.D. Htert promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer 2010, 10, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, Y.B.; Pirrotta, V. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Tolhuis, B.; de Wit, E.; Muijrers, I.; Teunissen, H.; Talhout, W.; van Steensel, B.; van Lohuizen, M. Genome-wide profiling of PRC1 and PRC2 polycomb chromatin binding in drosophila melanogaster. Nat. Genet. 2006, 38, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Del Mar Pena, L.M.; Laimins, L.A. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J. Virol. 2001, 75, 10005–10013. [Google Scholar] [CrossRef] [PubMed]
- Stunkel, W.; Bernard, H.U. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J. Virol. 1999, 73, 1918–1930. [Google Scholar] [PubMed]
- Swindle, C.S.; Engler, J.A. Association of the human papillomavirus type 11 E1 protein with histone H1. J. Virol. 1998, 72, 1994–2001. [Google Scholar] [PubMed]
- Wooldridge, T.R.; Laimins, L.A. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology 2008, 374, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. The binding of histone deacetylases and the integrity of Zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J. Virol. 2004, 78, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the hpv E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 2003, 22, 7871–7881. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.; Huh, K.W.; Munger, K. Human papillomavirus E7 oncoprotein dysregulates steroid receptor coactivator 1 localization and function. J. Virol. 2006, 80, 6669–6677. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; McCance, D.J. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on creb binding protein/p300 and P/CAF. J. Virol. 2002, 76, 8710–8721. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef] [PubMed]
- Hyland, P.L.; McDade, S.S.; McCloskey, R.; Dickson, G.J.; Arthur, K.; McCance, D.J.; Patel, D. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J. Virol. 2011, 85, 10999–11006. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co- activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- Jansma, A.L.; Martinez-Yamout, M.A.; Liao, R.; Sun, P.; Dyson, H.J.; Wright, P.E. The high-risk HPV16 E7 oncoprotein mediates interaction between the transcriptional coactivator CBP and the retinoblastoma protein prb. J. Mol. Biol. 2014, 426, 4030–4048. [Google Scholar] [CrossRef] [PubMed]
- Avantaggiati, M.L.; Ogryzko, V.; Gardner, K.; Giordano, A.; Levine, A.S.; Kelly, K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 1997, 89, 1175–1184. [Google Scholar] [CrossRef]
- Gu, W.; Shi, X.L.; Roeder, R.G. Synergistic activation of transcription by CBP and p53. Nature 1997, 387, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Felzien, L.K.; Betts, J.C.; Leung, K.; Beach, D.H.; Nabel, G.J. Regulation of NF-κB by cyclin-dependent kinases associated with the p300 coactivator. Science 1997, 275, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Snowden, A.W.; Perkins, N.D. Cell cycle regulation of the transcriptional coactivators p300 and creb binding protein. Biochem. Pharmacol. 1998, 55, 1947–1954. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. P300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Zhang, J.; Ellis, L.M.; Semenza, G.L.; Evans, D.B.; Watowich, S.S.; Gallick, G.E. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005, 24, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Chiang, C.M. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell 2005, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Cote, J.; Wang, W. Nurd, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 1998, 2, 851–861. [Google Scholar] [CrossRef]
- Zhang, Y.; LeRoy, G.; Seelig, H.P.; Lane, W.S.; Reinberg, D. The dermatomyositis-specific autoantigen MI2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 1998, 95, 279–289. [Google Scholar] [CrossRef]
- Longworth, M.S.; Wilson, R.; Laimins, L.A. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with hdacs. EMBO J. 2005, 24, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Um, S.J.; Rhyu, J.W.; Kim, E.J.; Jeon, K.C.; Hwang, E.S.; Park, J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Trojer, P.; Cao, A.R.; Gao, Z.; Li, Y.; Zhang, J.; Xu, X.; Li, G.; Losson, R.; Erdjument-Bromage, H.; Tempst, P.; et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol. Cell 2011, 42, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Whyte, W.A.; Anderssen, E.; Apostolou, E.; Chen, H.H.; Akbarian, S.; Bronson, R.T.; Hochedlinger, K.; Ramaswamy, S.; Young, R.A.; et al. The polycomb group protein L3MBTL2 assembles an atypical PRC1-family complex that is essential in pluripotent stem cells and early development. Cell Stem Cell 2012, 11, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic regulation of skin: Focus on the polycomb complex. Cell. Mol. Life Sci. 2012, 69, 2161–2172. [Google Scholar] [PubMed]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb group proteins are key regulators of keratinocyte function. J. Investig. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.; Martin, P. Epigenetic reprogramming during wound healing: Loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep. 2009, 10, 881–886. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Biochemical and functional interactions of human papillomavirus proteins with polycomb group proteins. Viruses 2013, 5, 1231–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, A.; Magnuson, T. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 1997, 13, 167–170. [Google Scholar] [CrossRef]
- Gould, A. Functions of mammalian Polycomb group and trithorax group related genes. Curr. Opin. Genet. Dev. 1997, 7, 488–494. [Google Scholar] [CrossRef]
- Hung, Y.C.; Ueda, M.; Terai, Y.; Kumagai, K.; Ueki, K.; Kanda, K.; Yamaguchi, H.; Akise, D.; Ueki, M. Homeobox gene expression and mutation in cervical carcinoma cells. Cancer Sci. 2003, 94, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Alami, Y.; Castronovo, V.; Belotti, D.; Flagiello, D.; Clausse, N. HOXC5 and HOXC8 expression are selectively turned on in human cervical cancer cells compared to normal keratinocytes. Biochem. Biophys. Res. Commun. 1999, 257, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Barba-de la Rosa, A.P.; Briones-Cerecero, E.; Lugo-Melchor, O.; De Leon-Rodriguez, A.; Santos, L.; Castelo-Ruelas, J.; Valdivia, A.; Pina, P.; Chagolla-Lopez, A.; Hernandez-Cueto, D.; et al. HOX B4 as potential marker of non-differentiated cells in human cervical cancer cells. J. Cancer Res. Clin. Oncol. 2012, 138, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Kuick, R.; Nan, B.; Ota, I.; Weiss, S.J.; Trimble, C.L.; Fearon, E.R.; Cho, K.R. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007, 67, 10163–10172. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.; Hoppe-Seyler, K.; Schuller, B.; Lohrey, C.; Maroldt, J.; Durst, M.; Hoppe-Seyler, F. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res. 2008, 68, 9964–9972. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. AKT-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27ME3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes. Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenfuhr, M.; Maertens, G.; Banck, M.; Zhou, M.M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4A/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and P16INK4A. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Sano, T.; Oyama, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. Expression status of p16 protein is associated with human papillomavirus oncogenic potential in cervical and genital lesions. Am. J. Pathol. 1998, 153, 1741–1748. [Google Scholar] [CrossRef]
- Klaes, R.; Friedrich, T.; Spitkovsky, D.; Ridder, R.; Rudy, W.; Petry, U.; Dallenbach-Hellweg, G.; Schmidt, D.; von Knebel Doeberitz, M. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int. J. Cancer 2001, 92, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Peng, K.L.; Jhang, H.C.; Lin, C.H.; Wu, S.Y.; Chiang, C.M.; Lee, S.C.; Yu, W.C.; Juan, L.J. The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 2012, 31, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Chin, H.G.; Esteve, P.O.; Jacobsen, S.E. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics 2009, 4, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Florence, B.; Faller, D.V. You BET-Cha: A novel family of transcriptional regulators. Front. Biosci. 2001, 6, D1008–D1018. [Google Scholar] [PubMed]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFB and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Jang, M.K. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 2013, 5, 1374–1394. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to hpv transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Olejnik-Schmidt, A.K.; Schmidt, M.T.; Kedzia, W.; Gozdzicka-Jozefiak, A. Search for cellular partners of human papillomavirus type 16 E2 protein. Arch. Virol. 2008, 153, 983–990. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Ozato, K.; McBride, A.A. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 2005, 79, 8920–8932. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.K.; McPhillips, M.G.; Ozato, K.; McBride, A.A. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 2005, 79, 4806–4818. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Anderson, D.E.; van Doorslaer, K.; McBride, A.A. A proteomic approach to discover and compare interacting partners of papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics 2015, 15, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the Brd4-positive transcription elongation factor b complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, V.K.; Zhang, Y.; Rajagopalan, D.; Abdullah, L.N.; Yeo-Teh, N.S.; Tomaic, V.; Banks, L.; Myers, M.P.; Chow, E.K.; Jha, S. E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60. Oncogene 2016, 35, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Pal Bhadra, M.; Girschick, H.J.; Bhadra, U. MicroRNAs—Micro in size but macro in function. FEBS J. 2008, 275, 4929–4944. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Li, G.; Laimins, L.A.; Cullen, B.R. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J. Virol. 2006, 80, 10890–10893. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. MicroRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.; Laimins, L.A. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J. Virol. 2013, 87, 6037–6043. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulation of microRNA-mRNA target pairs by human papillomavirus 16 oncoproteins. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Gardiner, A.S.; Board, K.F.; Monzon, F.A.; Edwards, R.P.; Khan, S.A. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 2008, 27, 2575–2582. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.M.; Marques, J.P.; Soares, A.R.; Carreto, L.; Santos, M.A. MicroRNA expression variability in human cervical tissues. PLoS ONE 2010, 5, e11780. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Tsang, T.Y.; Yau, P.L.; Kwok, T.T. Human papillomavirus type 16 E6 induces cervical cancer cell migration through the p53/microRNA-23b/urokinase-type plasminogen activator pathway. Oncogene 2011, 30, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Schwarz, J.K.; Lewis, J.S., Jr.; Huettner, P.C.; Rader, J.S.; Deasy, J.O.; Grigsby, P.W.; Wang, X. A microRNA expression signature for cervical cancer prognosis. Cancer Res. 2010, 70, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Wilting, S.M.; van Boerdonk, R.A.; Henken, F.E.; Meijer, C.J.; Diosdado, B.; Meijer, G.A.; le Sage, C.; Agami, R.; Snijders, P.J.; Steenbergen, R.D. Methylation-mediated silencing and tumour suppressive function of HSA-MIR-124 in cervical cancer. Mol. Cancer 2010, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Lui, W.O.; Pourmand, N.; Patterson, B.K.; Fire, A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007, 67, 6031–6043. [Google Scholar] [CrossRef] [PubMed]
- Patron, J.P.; Fendler, A.; Bild, M.; Jung, U.; Muller, H.; Arntzen, M.O.; Piso, C.; Stephan, C.; Thiede, B.; Mollenkopf, H.J.; et al. MIR-133b targets antiapoptotic genes and enhances death receptor-induced apoptosis. PLoS ONE 2012, 7, e35345. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.Q.; Wang, X.H.; Hou, L.J.; Jia, W.H.; Yang, Q.; Li, Y.X.; Liu, M.; Li, X.; Tang, H. MicroRNA-372 is down-regulated and targets cyclin-dependent kinase 2 (CDK2) and cyclin a1 in human cervical cancer, which may contribute to tumorigenesis. J. Biol. Chem. 2011, 286, 25556–25563. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Wang, X.B.; Chen, M.M.; Liu, T.; Li, Y.X.; Jia, W.H.; Liu, M.; Li, X.; Tang, H. MicroRNA-19a and -19b regulate cervical carcinoma cell proliferation and invasion by targeting CUL5. Cancer Lett. 2012, 322, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Davis, J.W.; Taylor, K.H.; Johnson, J.; Shi, Z.; Williams, R.; Atasoy, U.; Lewis, J.S., Jr.; Stack, M.S. Identification of a human papillomavirus-associated oncogenic miRNA panel in human oropharyngeal squamous cell carcinoma validated by bioinformatics analysis of the cancer genome atlas. Am. J. Pathol. 2015, 185, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Munger, K. Human papillomavirus 16 E6 and E7 oncoprotein expression alters microRNA expression in extracellular vesicles. Virology 2017, 508, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Lin, Z. MIR-21 is involved in cervical squamous cell tumorigenesis and regulates CCL20. Biochim. Biophys. Acta 2012, 1822, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta 2011, 1809, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Tsuchiya, S.; Meltzer, S.J.; Shimizu, K. MicroRNAs and epigenetics. FEBS J. 2011, 278, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R776. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, B.; Goldstein, L.D.; Ng, G.; Winder, D.M.; Palmer, R.D.; Gooding, E.L.; Barbosa-Morais, N.L.; Mukherjee, G.; Thorne, N.P.; Roberts, I.; et al. Global microRNA profiles in cervical squamous cell carcinoma depend on Drosha expression levels. J. Pathol. 2007, 212, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Subramaniyam, S.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; Schneider, A.; Arias-Pulido, H.; et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol. Cancer 2008, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Munger, K. Perturbation of drosha and dicer expression by human papillomavirus 16 oncoproteins. Virology 2017, 507, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Z.; Mei, Q.; Li, X.; Guo, M.; Fu, X.; Han, W. Long non-coding RNA HOTAIR, a driver of malignancy, predicts negative prognosis and exhibits oncogenic activity in oesophageal squamous cell carcinoma. Br. J. Cancer 2013, 109, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Rinn, J.L. Large non-coding RNAs: Missing links in cancer? Hum. Mol. Genet. 2010, 19, R152–R161. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Wang, X.L.; Tang, H.M.; Jiang, T.; Chen, J.; Lu, S.; Qiu, G.Q.; Peng, Z.H.; Yan, D.W. Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol. Rep. 2014, 32, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Kogo, R.; Shibata, K.; Sawada, G.; Takahashi, Y.; Kurashige, J.; Akiyoshi, S.; Sasaki, S.; Iwaya, T.; Sudo, T.; et al. Clinical significance of the expression of long non-coding RNA HOTAIR in primary hepatocellular carcinoma. Oncol. Rep. 2013, 29, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mandal, P.; Sadhukhan, T.; Roy Chowdhury, R.; Ranjan Mondal, N.; Chakravarty, B.; Chatterjee, T.; Roy, S.; Sengupta, S. Bridging links between long noncoding RNA HOTAIR and HPV oncoprotein E7 in cervical cancer pathogenesis. Sci. Rep. 2015, 5, 11724. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto, D.; Song, C.; McLaughlin-Drubin, M.E. Epigenetic Alterations in Human Papillomavirus-Associated Cancers. Viruses 2017, 9, 248. https://doi.org/10.3390/v9090248
Soto D, Song C, McLaughlin-Drubin ME. Epigenetic Alterations in Human Papillomavirus-Associated Cancers. Viruses. 2017; 9(9):248. https://doi.org/10.3390/v9090248
Chicago/Turabian StyleSoto, David, Christine Song, and Margaret E. McLaughlin-Drubin. 2017. "Epigenetic Alterations in Human Papillomavirus-Associated Cancers" Viruses 9, no. 9: 248. https://doi.org/10.3390/v9090248
APA StyleSoto, D., Song, C., & McLaughlin-Drubin, M. E. (2017). Epigenetic Alterations in Human Papillomavirus-Associated Cancers. Viruses, 9(9), 248. https://doi.org/10.3390/v9090248