
Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response

{kind=link}
{kind=link}
Abstract
1. Introduction
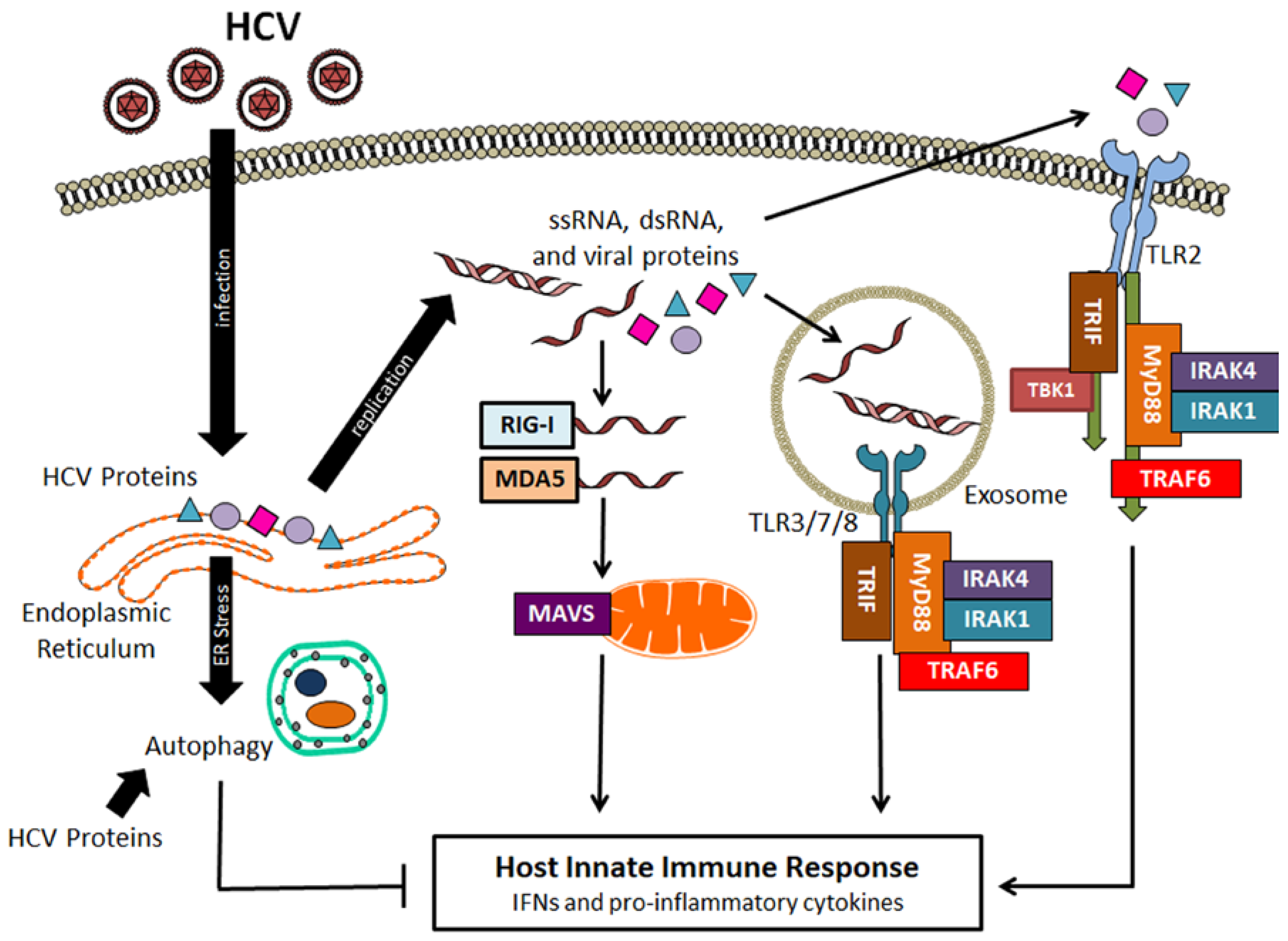
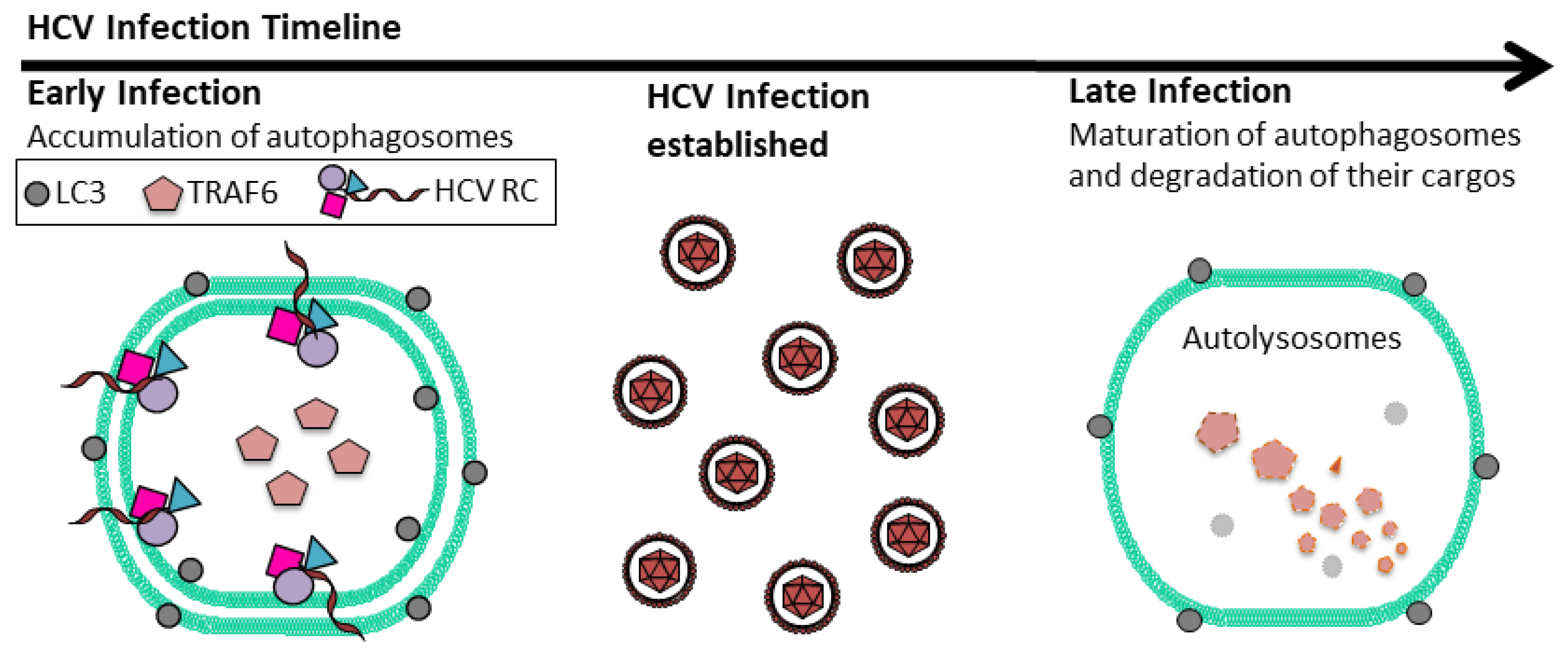
2. HCV and Autophagy
3. Autophagy on HCV Replication
4. Suppression of Host Innate Immune Response by HCV-Induced Autophagy
5. Suppression of HCV Replication by IFNs via Autophagy
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis c virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Marrone, A.; Restivo, L.; Guerrera, B.; Sellitto, A.; Rinaldi, L.; Romano, C.; Adinolfi, L.E. Chronic HCV infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J. Hepatol. 2013, 5, 528–540. [Google Scholar] [PubMed]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, N.L.; Wei, D.; Pfeffer, S.R.; Fan, M.; Pfeffer, L.M. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 2012, 55, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, Y.; Devaraj, S.; Wang, J.; Lemon, S.M.; Li, K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 2009, 83, 9824–9834. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Kodys, K.; Kopasz, A.; Marshall, C.; Do, T.; Romics, L., Jr.; Mandrekar, P.; Zapp, M.; Szabo, G. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J. Immunol. 2003, 170, 5615–5624. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Tian, Y.; Chan, S.T.; Kim, J.Y.; Cho, C.; Ou, J.H. TNF-α induced by hepatitis C virus via TLR7 and TLR8 in hepatocytes supports interferon signaling via an autocrine mechanism. PLoS Pathog. 2015, 11, e1004937. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Signal integration via pkr. Sci. STKE 2001, 2001, re2. [Google Scholar] [CrossRef] [PubMed]
- Toroney, R.; Nallagatla, S.R.; Boyer, J.A.; Cameron, C.E.; Bevilacqua, P.C. Regulation of PKR by HCV IRES RNA: Importance of domain II and NS5A. J. Mol. Biol. 2010, 400, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Shimoike, T.; McKenna, S.A.; Lindhout, D.A.; Puglisi, J.D. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antivir. Res. 2009, 83, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Dabo, S.; Meurs, E.F. Dsrna-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 2012, 4, 2598–2635. [Google Scholar] [CrossRef] [PubMed]
- Vegna, S.; Gregoire, D.; Moreau, M.; Lassus, P.; Durantel, D.; Assenat, E.; Hibner, U.; Simonin, Y. NOD1 participates in the innate immune response triggered by hepatitis C virus polymerase. J. Virol. 2016, 90, 6022–6035. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.T.; Chen, S.S. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell. Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef] [PubMed]
- Bauckman, K.A.; Owusu-Boaitey, N.; Mysorekar, I.U. Selective autophagy: Xenophagy. Methods 2015, 75, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; Kumar, D. Selective autophagy gets more selective: Uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis-infected macrophages. Autophagy 2016, 12, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W., 4th; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bieche, I.; Lebrec, D.; et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ou, J.H. Hepatitis C virus and autophagy. Biol. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Jheng, J.R.; Ho, J.Y.; Horng, J.T. ER stress, autophagy, and RNA viruses. Front. Microbiol. 2014, 5, 388. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand rna virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ou, J.H. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G847–G851. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Ploen, D.; Spengler, C.; Elgner, F.; Ren, H.; Bunten, S.; Hildt, E. HCV-induced oxidative stress by inhibition of NRF2 triggers autophagy and favors release of viral particles. Free Radic. Biol. Med. 2017, 110, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Aweya, J.J.; Mak, T.M.; Lim, S.G.; Tan, Y.J. The p7 protein of the hepatitis C virus induces cell death differently from the influenza a virus viroporin M2. Virus Res. 2013, 172, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [PubMed]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mtor signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, ATG14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Westphal, W.; Wong, K.N.; Tan, I.; Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl. Acad. Sci. USA 2010, 107, 19338–19343. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, A.M.; Labonte, P. The autophagy elongation complex (ATG5–12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci. Rep. 2017, 7, 40351. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J. Virol. 2015, 90, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011, 121, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.T.; Lee, J.; Narula, M.; Ou, J.J. Suppression of host innate immune response by hepatitis C virus via induction of autophagic degradation of traf6. J. Virol. 2016, 90, 10928–10935. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type i but not type iii ifn signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Kurt, R.; Chandra, P.K.; Aboulnasr, F.; Panigrahi, R.; Ferraris, P.; Aydin, Y.; Reiss, K.; Wu, T.; Balart, L.A.; Dash, S. Chaperone-mediated autophagy targets IFNAR1 for lysosomal degradation in free fatty acid treated HCV cell culture. PLoS ONE 2015, 10, e0125962. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.M.; Gong, B.; Chan, T.; Davey, R.A.; Soong, L.; Kolokoltsov, A.A.; Sun, J. Differential, type I interferon-mediated autophagic trafficking of hepatitis C virus proteins in mouse liver. Gastroenterology 2011, 141, 674–685, 685.e1–685.e6. [Google Scholar] [CrossRef] [PubMed]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein scotin interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Renzing, J.; Robertson, P.L.; Fernandes, K.N.; Lane, D.P. Scotin, a novel p53-inducible proapoptotic protein located in the er and the nuclear membrane. J. Cell Biol 2002, 158, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Fang, S.; Su, J.; Jiang, J.; Liang, B.; Huang, J.; Zhou, B.; Zang, N.; Ho, W.; et al. Downregulation of autophagy-related gene ATG5 and GABARAP expression by IFN-λ1 contributes to its anti-HCV activity in human hepatoma cells. Antivir. Res. 2017, 140, 83–94. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, S.T.; Ou, J.-h.J. Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses 2017, 9, 224. https://doi.org/10.3390/v9080224
Chan ST, Ou J-hJ. Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses. 2017; 9(8):224. https://doi.org/10.3390/v9080224
Chicago/Turabian StyleChan, Stephanie T., and Jing-hsiung James Ou. 2017. "Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response" Viruses 9, no. 8: 224. https://doi.org/10.3390/v9080224
APA StyleChan, S. T., & Ou, J.-h. J. (2017). Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses, 9(8), 224. https://doi.org/10.3390/v9080224