Distribution and Inferred Evolutionary Characteristics of a Chimeric ssDNA Virus Associated with Intertidal Marine Isopods


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isopod Collection and Virome Analysis
2.2. IWaV278 Genome Analysis and Quantitation
3. Results and Discussion
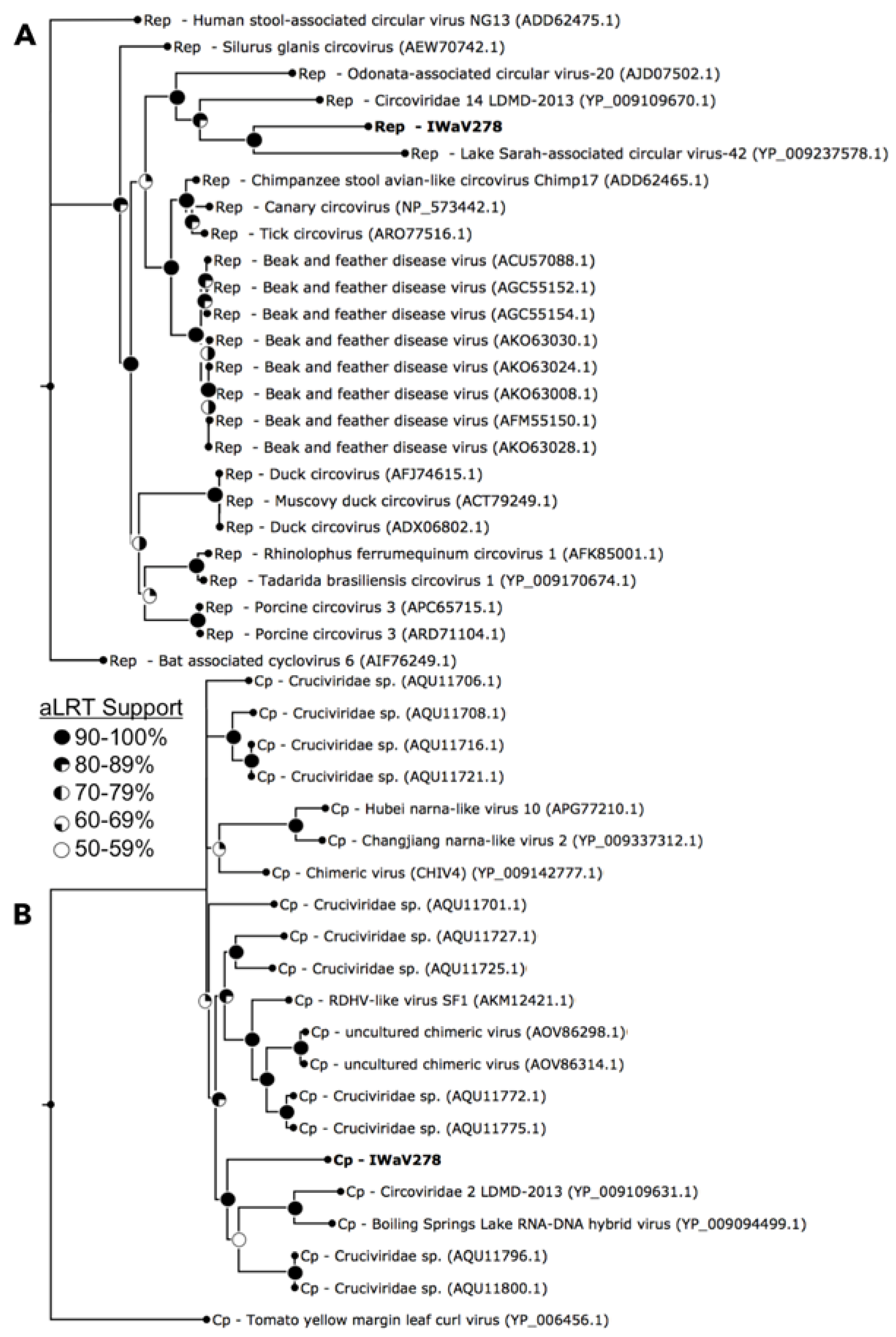
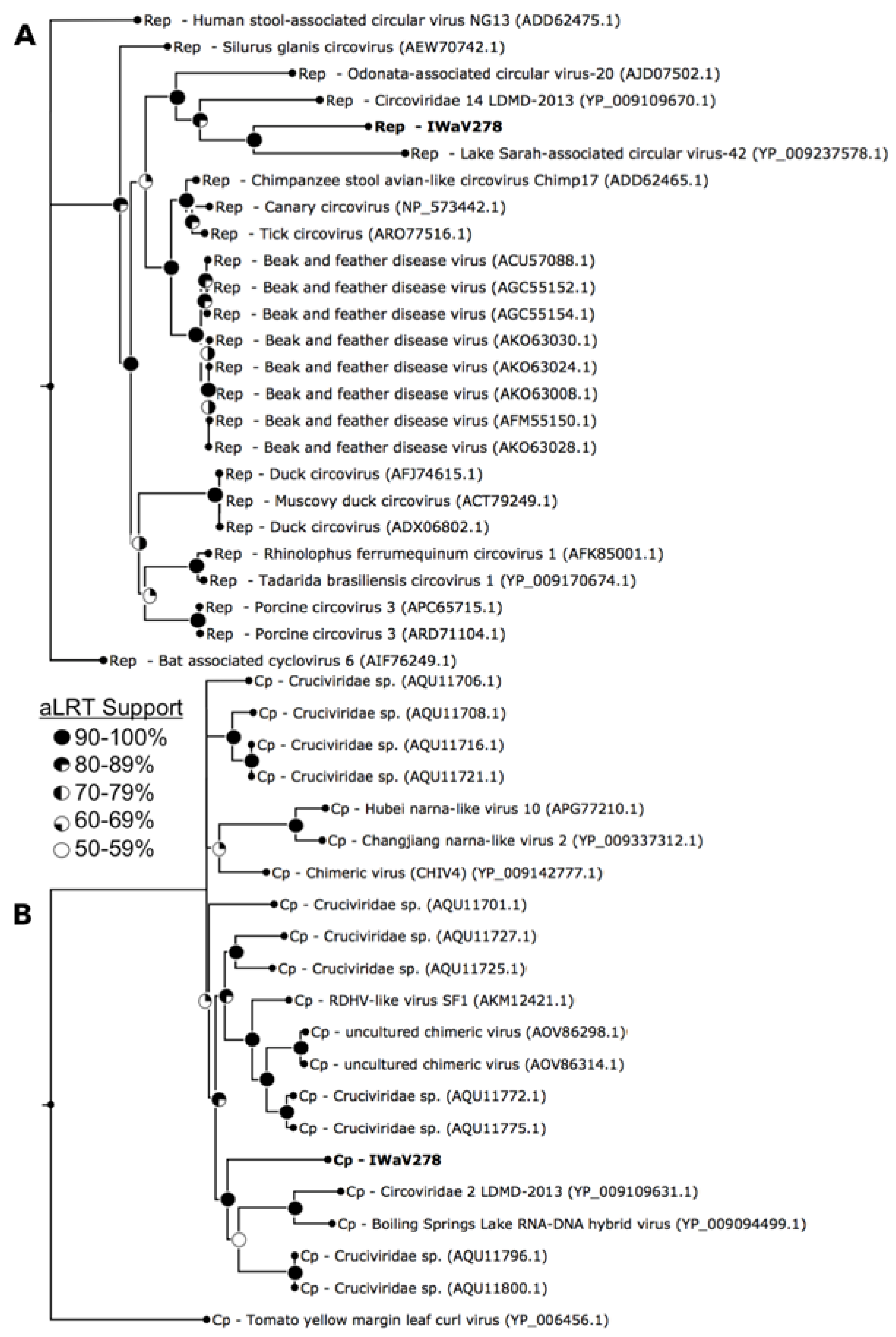
3.1. Identification of Isopod-Associated CRESS-DNA and Chimeric Viruses
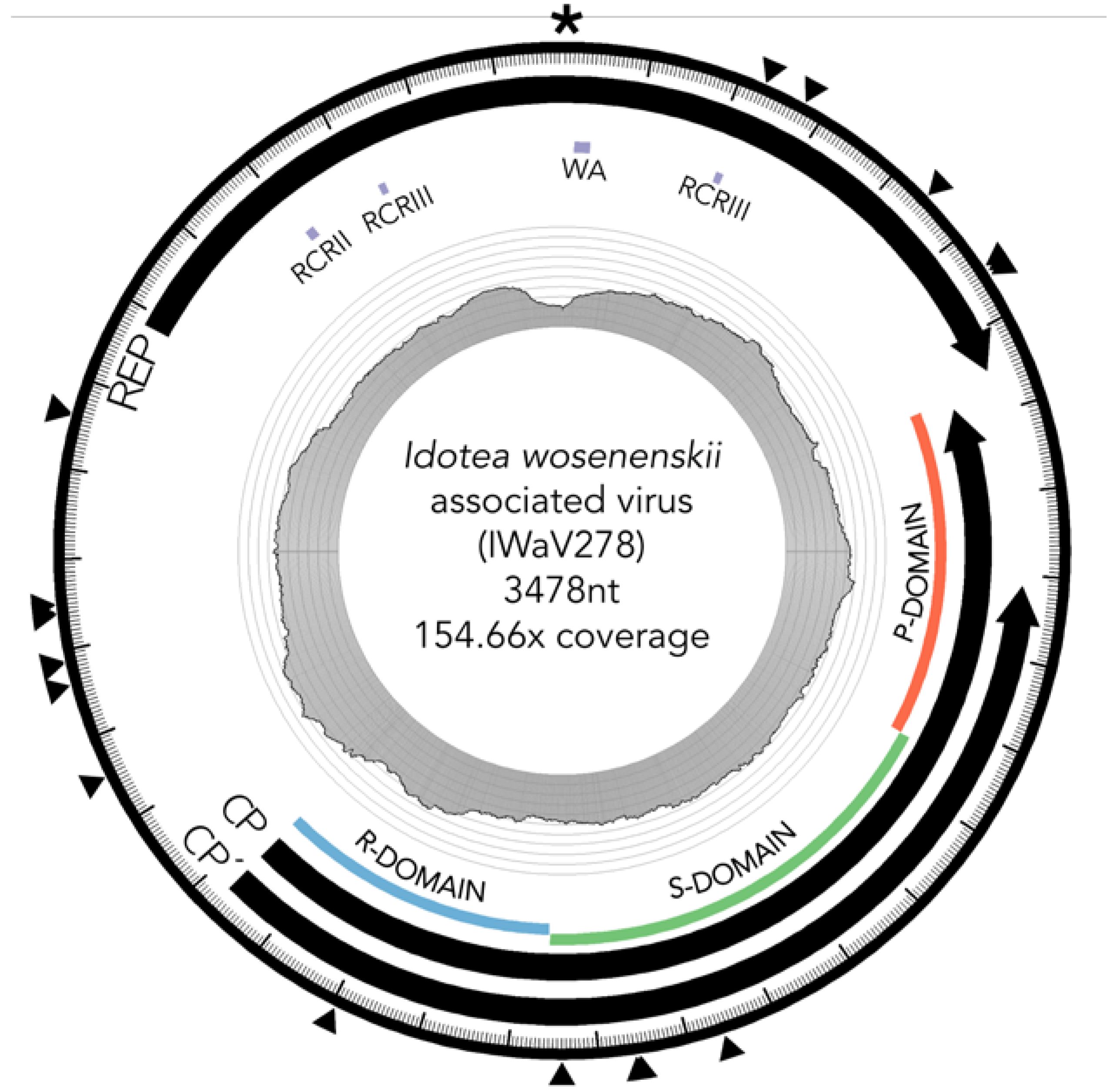
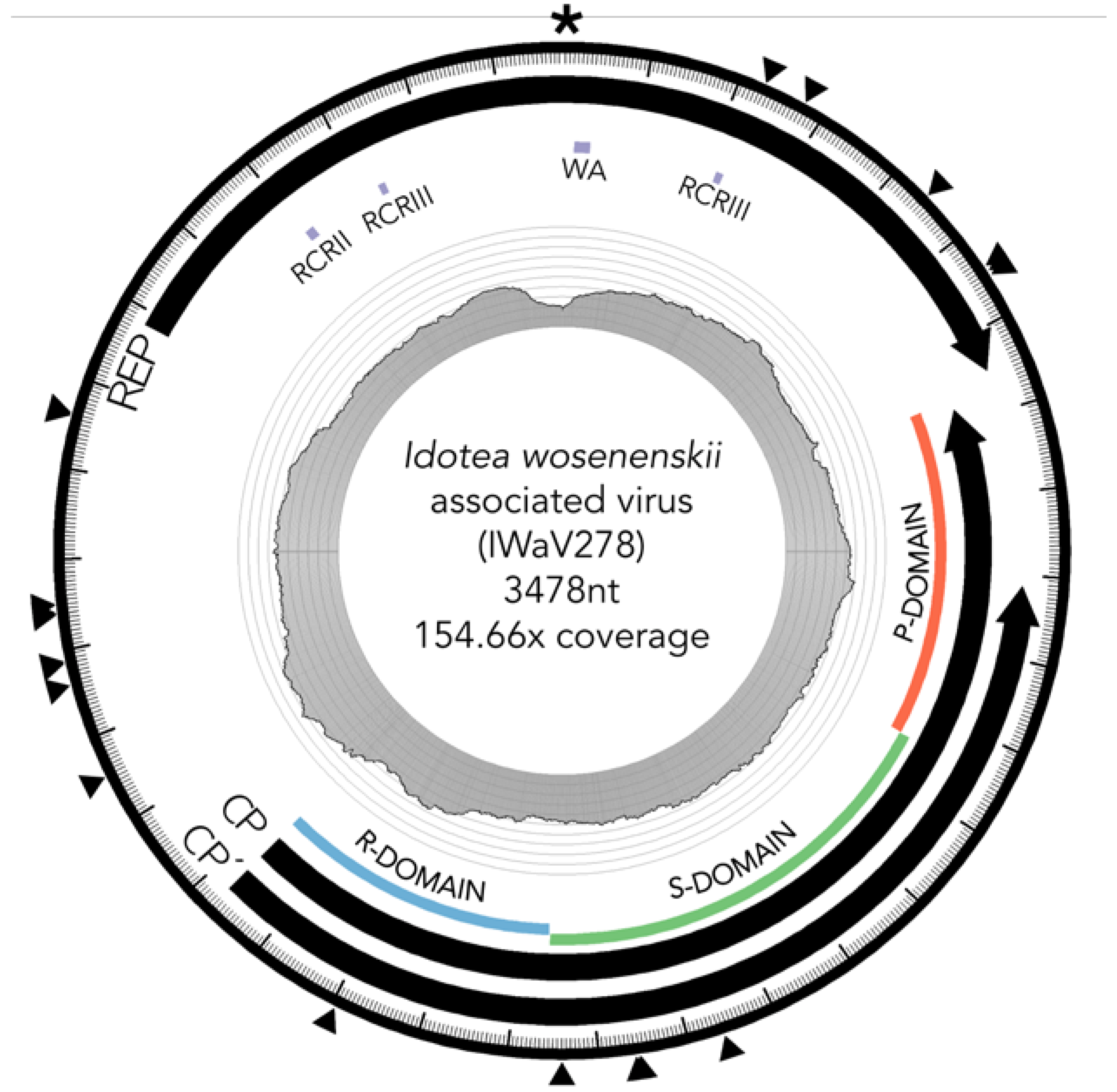
3.2. Characterization of I. wosnesenskii Associated Chimeric Virus IWaV278
3.2.1. Replication Initiation ORF (IWaV278-Rep)
3.2.2. Structural ORF (IWaV278-Cp)
3.3. IWaV278 Codon Usage Biases and Sequence Conservation
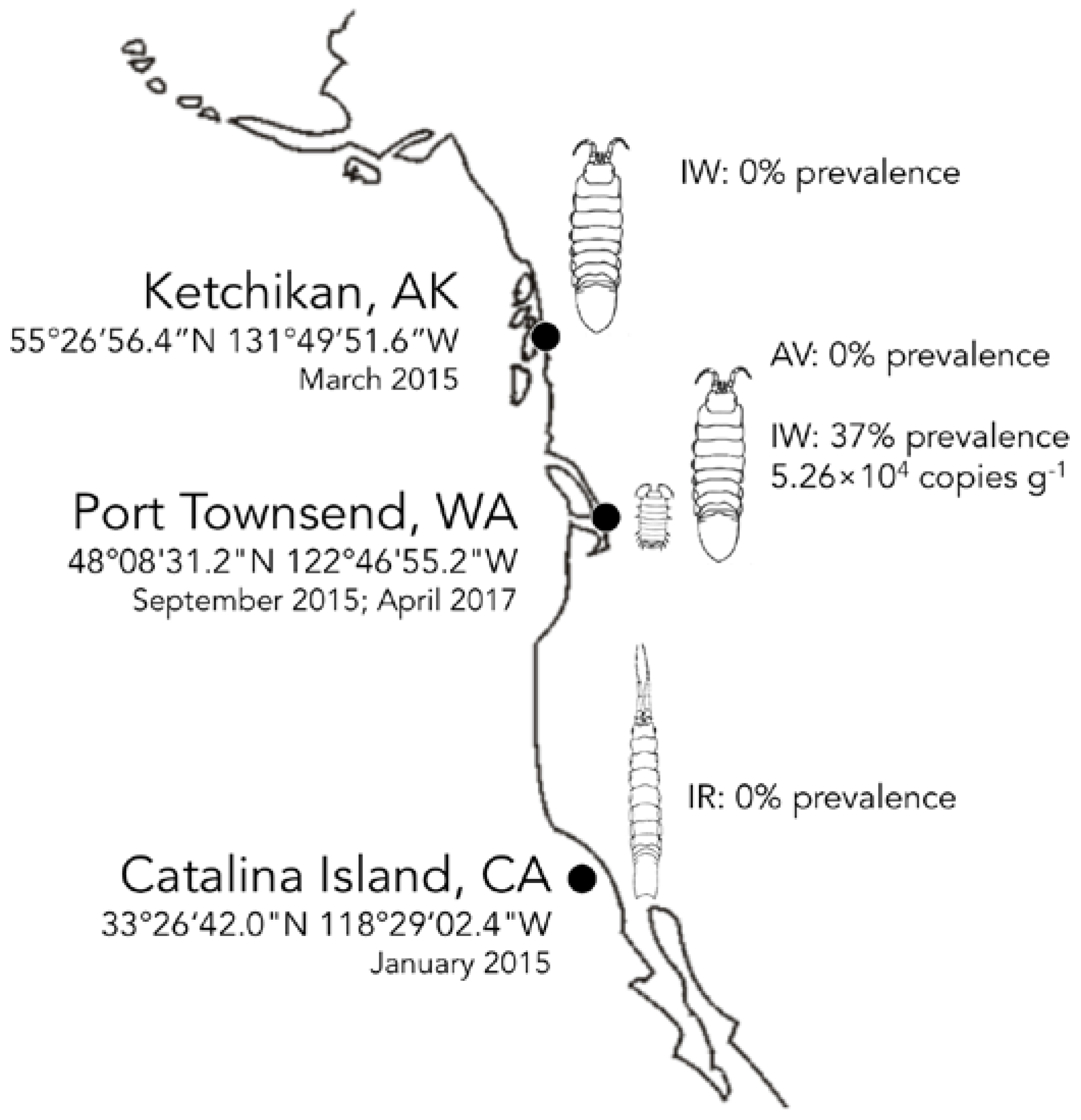
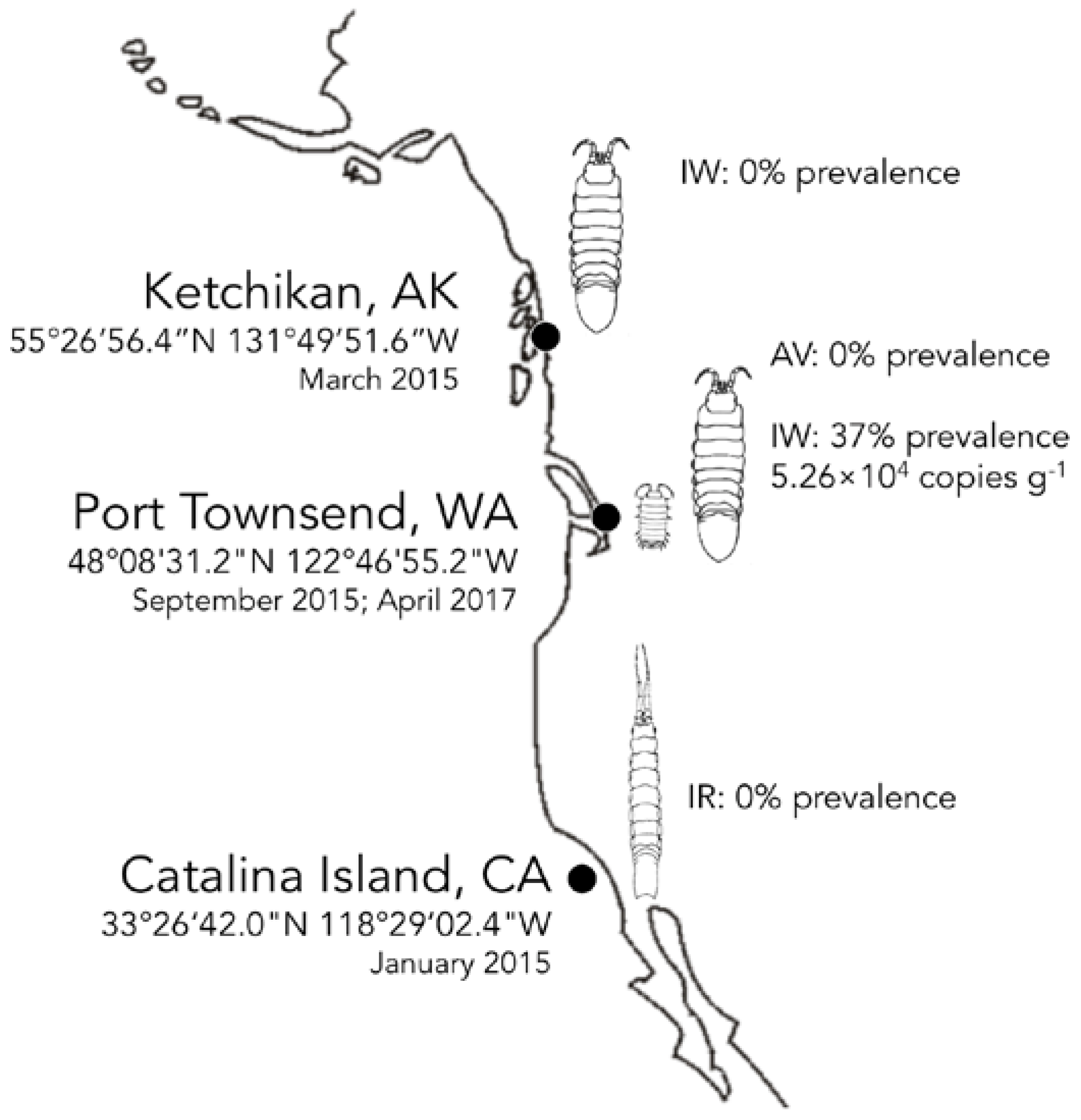
3.4. Biogeography and Transience of IWaV278
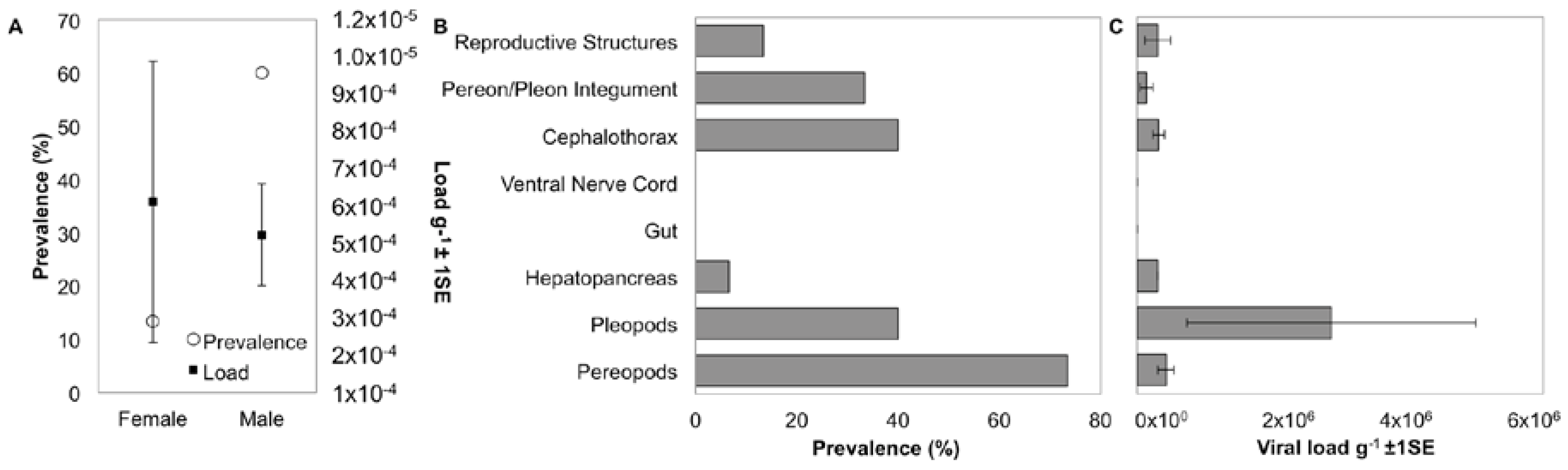
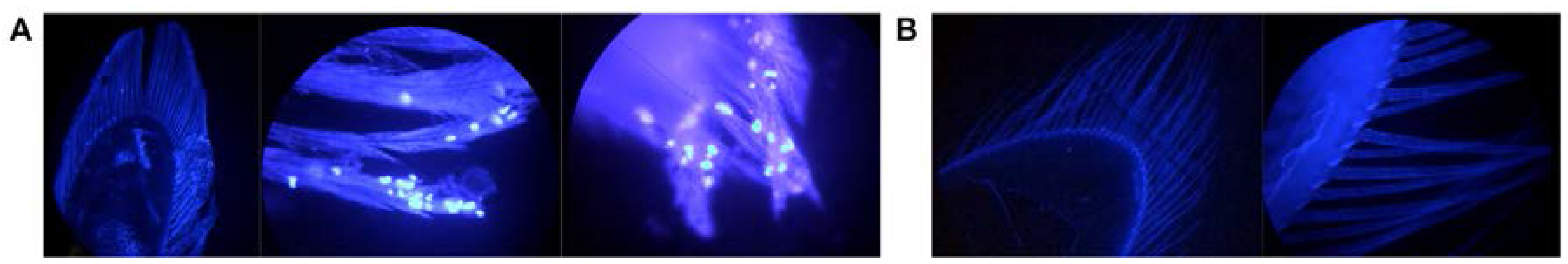
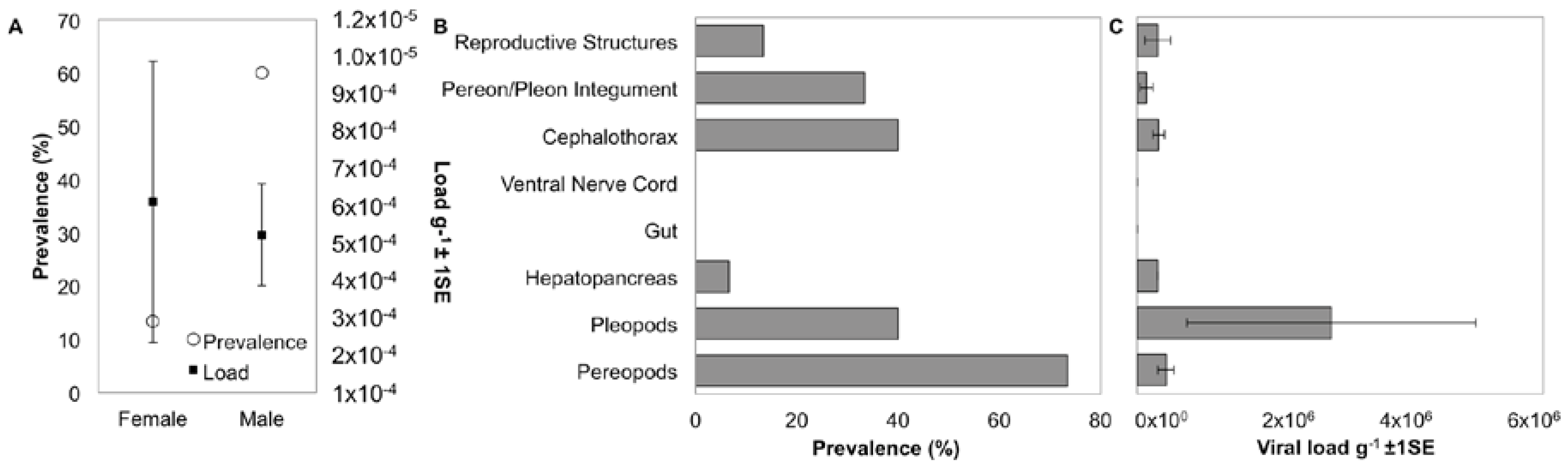
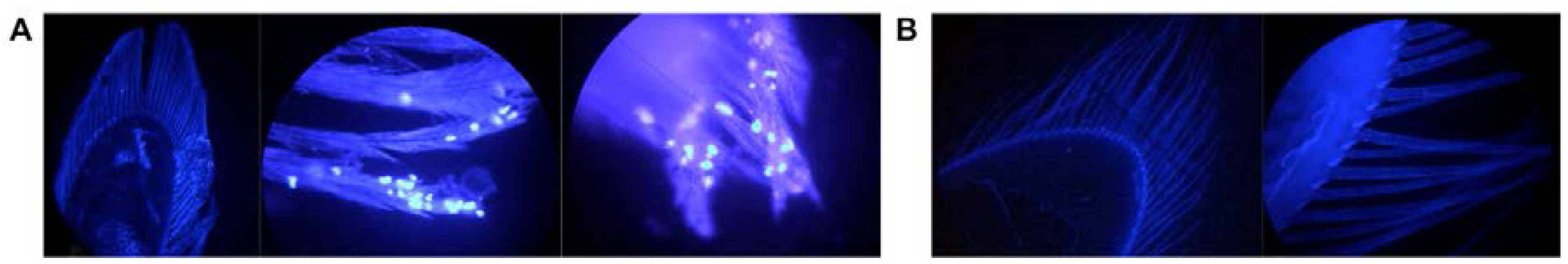
3.5. Localization and Predicted Host of IWaV278
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clark, D.R.; Aazem, K.V.; Hays, G.C. Zooplankton abundance and community structure over a 4,000 km transect in the northeast Atlantic. J. Plankton Res. 2001, 23, 365–372. [Google Scholar] [CrossRef]
- Martin, J.W.; Davis, G.E. An updated classification of the recent Crustacea. Q. Rev. Biol. 2003, 39, 1–124. [Google Scholar]
- Dole-Olivier, M.J.; Galassi, D.M.P.; Marmonier, P.; Creuze des Chatelliers, M. The biology and ecology of lotic microcrustaceans. Freshw. Biol. 2000, 44, 63–91. [Google Scholar] [CrossRef]
- Leidenberger, S.; Harding, K.; Jonsson, P.R. Ecology and distribution of the isopod genus Idotea in the Baltic Sea: Key species in a changing environment. J. Crustac. Biol. 2012, 32, 359–381. [Google Scholar] [CrossRef]
- Neill, W.E. Experimental studies of microcrustacean competition, community composition and efficiency of resource utilization. Ecology 1975, 56, 809–826. [Google Scholar] [CrossRef]
- Bistolas, K.S.I.; Jackson, E.W.; Watkins, J.M.; Rudstam, L.G.; Hewson, I. Distribution of circular single-stranded DNA viruses associated with benthic amphipods of genus Diporeia in the Laurentian Great Lakes. Freshw. Biol. 2017, 62, 1220–1231. [Google Scholar] [CrossRef]
- Dunlap, D.S.; Ng, T.F.F.; Rosario, K.; Barbosa, J.G.; Greco, A.M.; Breitbart, M.; Hewson, I. Molecular and microscopic evidence of viruses in marine copepods. Proc. Natl. Acad. Sci. USA 2013, 110, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Gudenkauf, B.M.; Hewson, I. Comparative metagenomics of viral assemblages inhabiting four phyla of marine invertebrates. Front. Mar. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Hewson, I.; Ng, G.; Li, W.; LaBarre, B.A.; Aguirre, I.; Barbosa, J.G.; Breitbart, M.; Greco, A.W.; Kearns, C.M.; Looi, A.; et al. Metagenomic identification, seasonal dynamics, and potential transmission mechanisms of a Daphnia-associated single-stranded DNA virus in two temperate lakes. Limnol. Oceanogr. 2013, 58, 1605–1620. [Google Scholar] [CrossRef]
- Rosario, K.; Schenck, R.O.; Harbeitner, R.C.; Lawler, S.N.; Breitbart, M. Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Eaglesham, J.; Hewson, I. Widespread detection of circular replication initiator protein (rep)-encoding ssDNA viral genomes in estuarine, coastal and open ocean net plankton. Mar. Ecol. Prog. Ser. 2013, 494, 65–72. [Google Scholar] [CrossRef]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. Diverse circovirus-like genome architectures revealed by environmental metagenomics. J. Gen. Virol. 2009, 90, 2418–2424. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Zhi, N.; Li, J.; Hu, G.; Koonin, E.V.; Wong, S.; Shevchenko, S.; Zhao, K.; Young, N.S. Multiple layers of chimerism in a single-stranded DNA virus discovered by deep sequencing. Genome Biol. Evol. 2015, 7, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.M.; Varsani, A.; Martin, D.P. Widely conserved recombination patterns among single-stranded DNA viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef] [PubMed]
- Stedman, K.M. Deep recombination: RNA and ssDNA virus genes in DNA virus and host genomes. Annu. Rev. Virol. 2015, 2, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Diemer, G.S.; Stedman, K.M. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol. Direct 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Pearson, V.M.; Caudle, S.B.; Rokyta, D.R. Viral recombination blurs taxonomic lines: Examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 2016, 4, e2585. [Google Scholar] [CrossRef] [PubMed]
- Quaiser, A.; Krupovic, M.; Dufresne, A.; Francez, A.J.; Roux, S. Diversity and comparative genomics of chimeric viruses in Sphagnum-dominated peatlands. Virus Evol. 2016, 2, vew025. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Bronner, G.; Vaulot, D.; Forterre, P.; Krupovic, M. Chimeric viruses blur the borders between the major groups of eukaryotic single-stranded DNA viruses. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, A.; Galatowitsch, M.L.; Argüello-Astorga, G.R.; van Bysterveldt, K.; Kraberger, S.; Stainton, D.; Harding, J.S.; Roumagnac, P.; Martin, D.P.; Lefeuvre, P.; et al. Diverse circular replication-associated protein encoding viruses circulating in invertebrates within a lake ecosystem. Infect. Genet. Evol. 2016, 39, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.D.; Rosario, K.; Breitbart, M.; Paul, J.H. Comparative metagenomics: Natural populations of induced prophages demonstrate highly unique, lower diversity viral sequences: Metagenomic comparison of induced and ambient viruses. Environ. Microbiol. 2014, 16, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M. Recombination between RNA viruses and plasmids might have played a central role in the origin and evolution of small DNA viruses. BioEssays 2012, 34, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.C.B.; Bruce, N.L. Global diversity of marine isopods (except Asellota and crustacean symbionts). PLoS ONE 2012, 7, e43529. [Google Scholar] [CrossRef]
- Eriksen, C.H. Aspects of the limno-ecology of Corophium spinicorne Stimpson (Amphipoda) and Gnorimosphaeroma oregonensis (Dana) (Isopoda). Crustaceana 1968, 14, 1–12. [Google Scholar] [CrossRef]
- Lee, W.L.; Gilchrist, B.M. Pigmentation, color change and the ecology of the marine isopod Idotea resecata (Stimpson). J. Exp. Mar. Biol. Ecol. 1972, 10, 1–27. [Google Scholar] [CrossRef]
- Naylor, E. The diet and feeding mechanism of Idotea. J. Mar. Biol. Assoc. UK 1955, 34, 347. [Google Scholar] [CrossRef]
- Ruesink, J.L. Intertidal mesograzers in field microcosms: Linking laboratory feeding rates to community dynamics. J. Exp. Mar. Biol. Ecol. 2000, 248, 163–176. [Google Scholar] [CrossRef]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O.; Sullivan, J. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Rudy, P.J.; Rudy, L.H.; Shanks, A.L.; Butler, B. Idotea wosnesenskii. In Oregon Estuarine Invertebrates: Rudys’ Illustrated Guide to Common Species; University of Oregon Libraries and Oregon Institute of Marine Biology: Charleston, OR, USA, 2015. [Google Scholar]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols; Humana Press: Clifton, NJ, USA, 1999; Volume 132, pp. 365–386. ISBN 978-1-59259-192-3. [Google Scholar]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the marine virosphere using metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Bae, J.W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Burgyan, J.; Martelli, G.P. Molecular biology of Tombusviridae. In Advances In Virus Research; Elsevier: Amsterdam, The Netherlands, 1994; Volume 44, pp. 381–428. ISBN 978-0-12-039844-7. [Google Scholar]
- Heller-Dohmen, M.; Göpfert, J.C.; Pfannstiel, J.; Spring, O. The nucleotide sequence and genome organization of Plasmopara halstedii virus. Virol. J. 2011, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, T.; Yamashita, S.; Hibi, T. The nucleotide sequence and genome organization of Sclerophthora macrospora virus A. Virology 2003, 311, 394–399. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 2006, 62, 551. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.A.; Chubb, J.C. Veltkamp Epibionts of Asellus aquaticus (L.) (Crustacea, Isopoda): An SEM study. Freshw. Biol. 1998, 39, 423–438. [Google Scholar] [CrossRef]
- Fernandez-Leborans, G.; Hanamura, Y.; Nagasaki, K. Presence of two new species of suctorian (protozoa: Ciliophora) epibiont on the marine crustacean isopod Excirolana chiltoni. J. Mar. Biol. Assoc. UK 2003, 23, 447–452. [Google Scholar] [CrossRef]
- Ólafsdóttir, S.H.; Svavarsson, J. Ciliate (protozoa) epibionts of deep-water Asellote isopods (Crustacea): Pattern and diversity. J. Crustac. Biol. 2002, 22, 607–618. [Google Scholar] [CrossRef]
- Kováčiková, M.; Simdyanov, T.G.; Diakin, A.; Valigurová, A. Structures related to attachment and motility in the marine eugregarine Cephaloidophora cf. communis (Apicomplexa). Eur. J. Protistol. 2017, 59, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.H. The small subunit rRNA gene sequence of the chonotrich Chilodochona carcini Jankowski, 1973 confirms chonotrichs as a dysteriid-derived clade (Phyllopharyngea, Ciliophora). Int. J. Syst. Evol. Microbiol. 2016, 66, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bistolas, K.S.I.; Besemer, R.M.; Rudstam, L.G.; Hewson, I. Distribution and Inferred Evolutionary Characteristics of a Chimeric ssDNA Virus Associated with Intertidal Marine Isopods. Viruses 2017, 9, 361. https://doi.org/10.3390/v9120361
Bistolas KSI, Besemer RM, Rudstam LG, Hewson I. Distribution and Inferred Evolutionary Characteristics of a Chimeric ssDNA Virus Associated with Intertidal Marine Isopods. Viruses. 2017; 9(12):361. https://doi.org/10.3390/v9120361
Chicago/Turabian StyleBistolas, Kalia S. I., Ryan M. Besemer, Lars G. Rudstam, and Ian Hewson. 2017. "Distribution and Inferred Evolutionary Characteristics of a Chimeric ssDNA Virus Associated with Intertidal Marine Isopods" Viruses 9, no. 12: 361. https://doi.org/10.3390/v9120361
APA StyleBistolas, K. S. I., Besemer, R. M., Rudstam, L. G., & Hewson, I. (2017). Distribution and Inferred Evolutionary Characteristics of a Chimeric ssDNA Virus Associated with Intertidal Marine Isopods. Viruses, 9(12), 361. https://doi.org/10.3390/v9120361