Autophagy Proteins in Viral Exocytosis and Anti-Viral Immune Responses


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
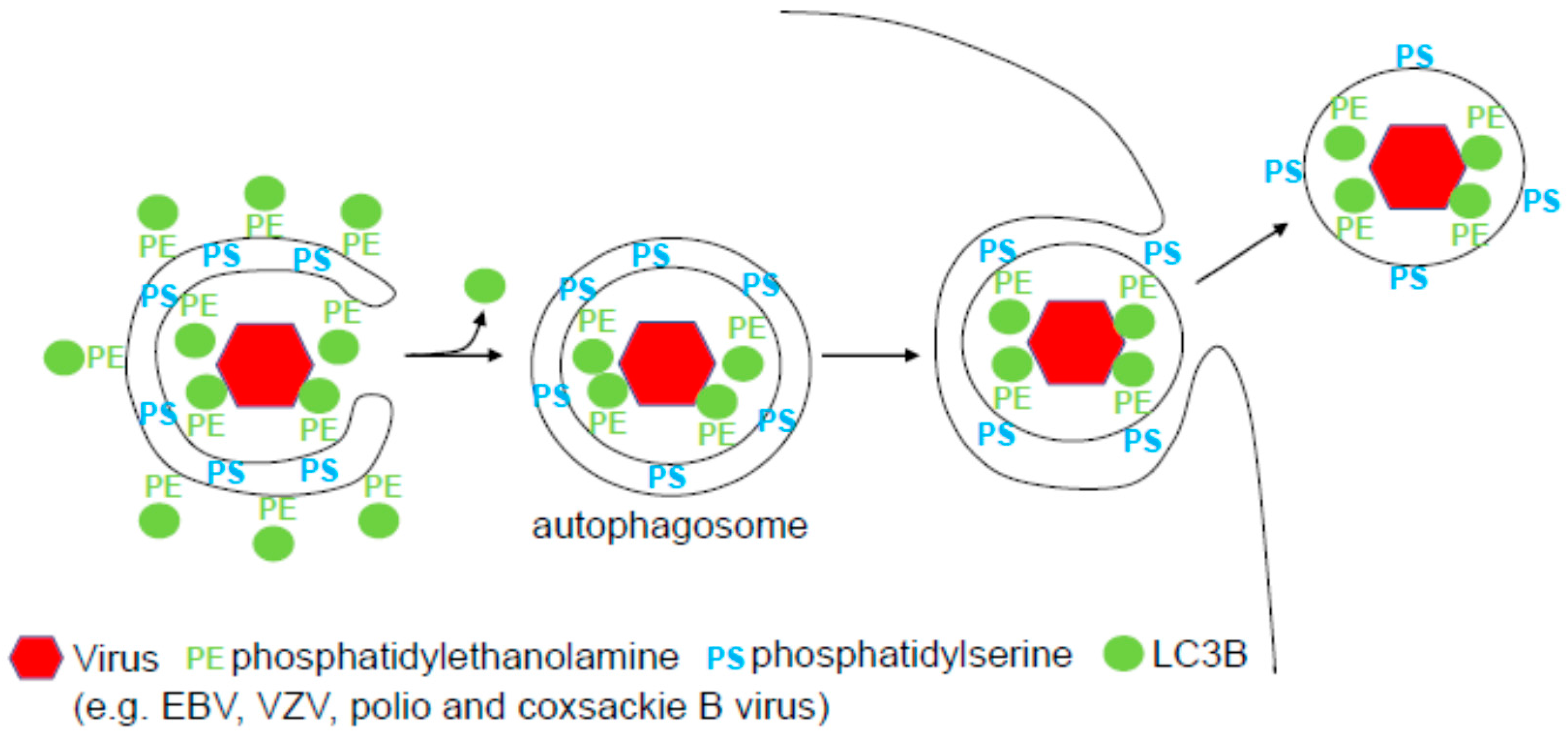
2. Autophagic Envelope Acquisition by Herpes Viruses
3. Atg Assisted Exocytosis of Picornaviruses
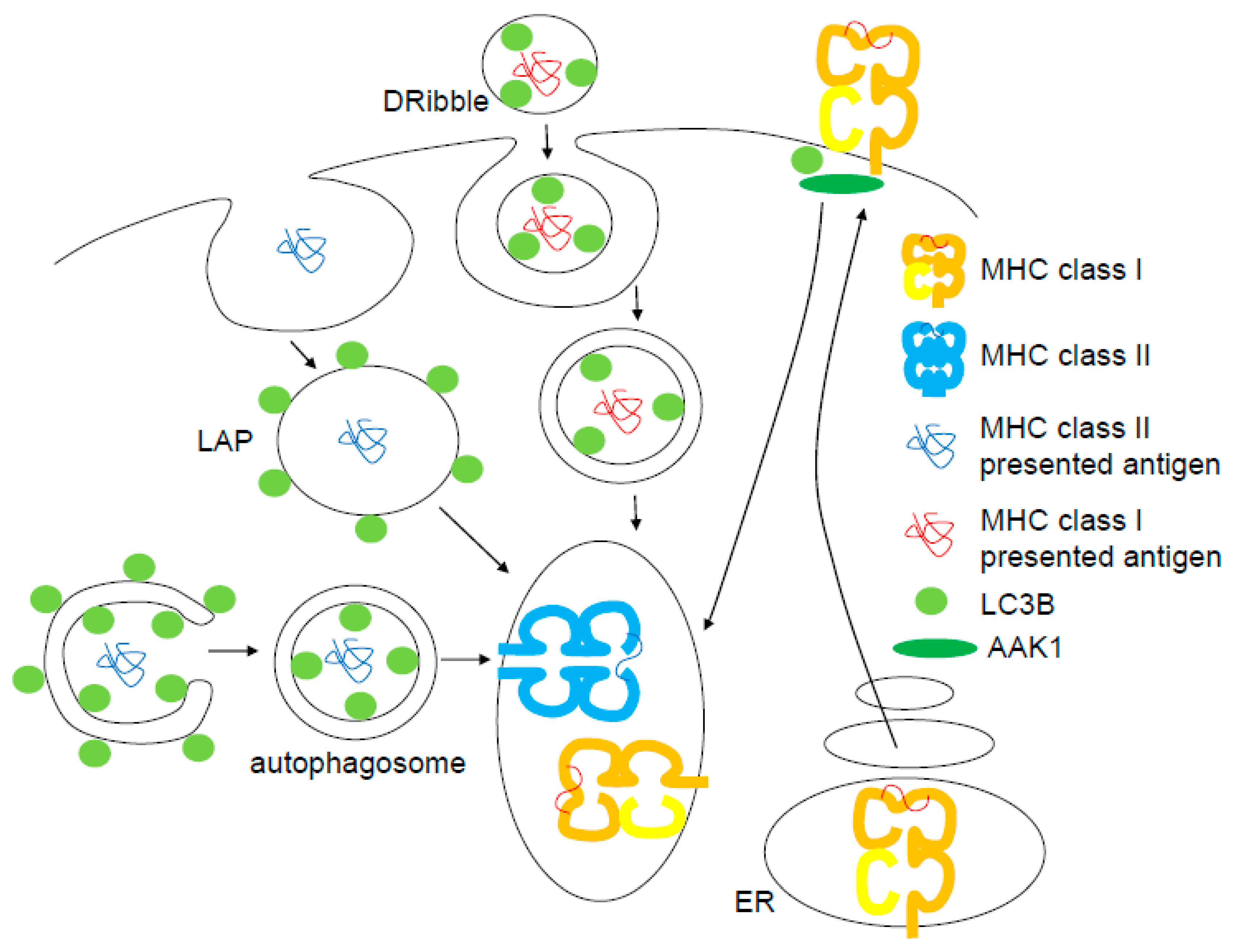
4. Exo- and Endocytosis with Atg Modules for MHC Class I Antigen Presentation
5. Autophagy and LAP for MHC Class II Antigen Presentation
6. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through hops complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored snare syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. The macroautophagy machinery in endo- and exocytosis. J. Mol. Biol. 2017, 429, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 α (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Henault, J.; Martinez, J.; Riggs, J.M.; Tian, J.; Mehta, P.; Clarke, L.; Sasai, M.; Latz, E.; Brinkmann, M.M.; Iwasaki, A.; et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 2012, 37, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Romao, S.; Gasser, N.; Becker, A.C.; Guhl, B.; Bagajic, M.; Vanoaica, L.D.; Ziegler, U.; Roesler, J.; Dengjel, J.; Reichenbach, J.; et al. Essential autophagy proteins stabilize pathogen containing phagosomes for prolonged MHC class II antigen processing. J. Cell Biol. 2013, 203, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Manjithaya, R.; Anjard, C.; Loomis, W.F.; Subramani, S. Unconventional secretion of pichia pastoris Acb1 is dependent on grasp protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010, 188, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated snares and specialized trim cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, D.; Curwin, A.J.; Popoff, J.F.; Bruns, C.; Duran, J.M.; Malhotra, V. Remodeling of secretory compartments creates cups during nutrient starvation. J. Cell Biol. 2014, 207, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, V. Unconventional protein secretion: An evolving mechanism. Embo J. 2013, 32, 1660–1664. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. The autophagic machinery in viral exocytosis. Front. Microbiol. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Nowag, H.; Guhl, B.; Thriene, K.; Romao, S.; Ziegler, U.; Dengjel, J.; Münz, C. Macroautopphagy proteins assist epstein barr virus production and get incorporated into the virus particles. EBioMedicine 2014, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Jarosinski, K.W.; Jackson, W.; Carpenter, J.E.; Grose, C. Exocytosis of varicella-zoster virions involves a convergence of endosomal and autophagy pathways. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. EBV blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and biological studies on a virus in cultured lymphoblasts from burkitt’s lymphoma. J. Exp. Med. 1964, 121, 761–770. [Google Scholar] [CrossRef]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron microscopic study of the formation of poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, A.M.; Labonte, P. The autophagy elongation complex (ATG5–12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci. Rep. 2017, 7, 40351. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Wang, L.; Lee, J.; Ou, J.J. HCV induces the localization of lipid rafts to autophagosomes for its RNA replication. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Biering, S.B.; Choi, J.; Halstrom, R.A.; Brown, H.M.; Beatty, W.L.; Lee, S.; McCune, B.T.; Dominici, E.; Williams, L.E.; Orchard, R.C.; et al. Viral replication complexes are targeted by LC3-guided interferon-inducible GTPases. Cell Host Microbe 2017, 22, 74–85.e7. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012, 8, e1003046. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- McKnight, K.L.; Xie, L.; Gonzalez-Lopez, O.; Rivera-Serrano, E.E.; Chen, X.; Lemon, S.M. Protein composition of the hepatitis A virus quasi-envelope. Proc. Natl. Acad. Sci. USA 2017, 114, 6587–6592. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Sirois, I.; Bell, C.; Hanafi, L.A.; Hamelin, K.; Dieude, M.; Rondeau, C.; Thibault, P.; Desjardins, M.; Hebert, M.J. A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics 2013, 13, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.X.; Yang, G.; Hao, F.; Urba, W.J.; Hu, H.M. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008, 68, 6889–6895. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Kepp, O.; Jusforgues-Saklani, H.; Vicencio, J.M.; Kroemer, G.; Albert, M.L. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009, 16, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Li, Y.; Cui, Z.; Morris, N.P.; Weinberg, A.D.; Fox, B.A.; Urba, W.J.; Wang, L.; Hu, H.M. Combinational immunotherapy with allo-dribble vaccines and anti-OX40 co-stimulation leads to generation of cross-reactive effector t cells and tumor regression. Sci. Rep. 2016, 6, 37558. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Muller, A.; Steinbach, K.; Niven, J.; Barreira da Silva, R.; Paul, P.; Ligeon, L.A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8+ T cell responses. Cell Rep. 2016, 15, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.W.; Loi, M.; Ewert, S.; Quast, I.; Theiler, R.; Gannage, M.; Münz, C.; de Libero, G.; Freigang, S.; Lunemann, J.D. The autophagy machinery restrains iNKT cell activation through CD1D1 internalization. Autophagy 2017, 13, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Hubbard-Lucey, V.M.; Shono, Y.; Maurer, K.; West, M.L.; Singer, N.V.; Ziegler, C.G.K.; Lezcano, C.; Motta, A.C.F.; Schmid, K.; Levi, S.M.; et al. Autophagy gene ATG16L1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity 2014, 41, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yokoyama, C.C.; Williams, J.W.; Baldridge, M.T.; Jin, X.; DesRochers, B.; Bricker, T.; Wilen, C.B.; Bagaitkar, J.; Loginicheva, E.; et al. Homeostatic control of innate lung inflammation by Vici syndrome gene EPG5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe 2016, 19, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.; Cacheux, W.; Bara, M.A.; L'Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Becker, C.; Lowell, C.A.; Underhill, D.M. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J. Biol. Chem. 2012, 287, 34149–34156. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Cunha, L.D.; Park, S.; Yang, M.; Lu, Q.; Orchard, R.; Li, Q.Z.; Yan, M.; Janke, L.; Guy, C.; et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016, 533, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Becker, C.; Reyes, C.; Underhill, D.M. Cutting edge: Fyco1 recruitment to Dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J. Immunol. 2014, 192, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Suri, A.; Walters, J.J.; Rohrs, H.W.; Gross, M.L.; Unanue, E.R. First signature of islet β-cell-derived naturally processed peptides selected by diabetogenic class II MHC molecules. J. Immunol. 2008, 180, 3849–3856. [Google Scholar] [CrossRef] [PubMed]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Aichinger, M.; Wu, C.; Nedjic, J.; Klein, L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J. Exp. Med. 2013, 210, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Kasai, M.; Tanida, I.; Ueno, T.; Kominami, E.; Seki, S.; Ikeda, T.; Mizuochi, T. Autophagic compartments gain access to the MHC class II compartments in thymic epithelium. J. Immunol. 2009, 183, 7278–7285. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, P.A.; Leib, D.A. Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 2012, 3, e00267-12. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.K.; Baena, A.; Ng, T.W.; Venkataswamy, M.M.; Kennedy, S.C.; Kunnath-Velayudhan, S.; Carreno, L.J.; Xu, J.; Chan, J.; Larsen, M.H.; et al. Suppression of autophagy and antigen presentation by mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016, 1, 16133. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. MHC class II antigen loading compartments continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Comber, J.D.; Robinson, T.M.; Siciliano, N.A.; Snook, A.E.; Eisenlohr, L.C. Functional macroautophagy induction by influenza a virus without a contribution to MHC-class II restricted presentation. J. Virol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.F.; Brilot, F.; Münz, C.; Gannage, M. The tumor antigen NY-ESO-1 mediates direct recognition of melanoma cells by CD4+ T cells after intercellular antigen transfer. J. Immunol. 2016, 196, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Coulon, P.G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-infected dendritic cells present endogenous MHC class II-restricted antigens to HIV-specific CD4+ T cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sun, C.; Feng, L.; Li, P.; Xiao, L.; Ren, Y.; Wang, D.; Li, C.; Chen, L. Regulation of SIV antigen-specific CD4+ T cellular immunity via autophagosome-mediated MHC II molecule-targeting antigen presentation in mice. PLoS ONE 2014, 9, e93143. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Münz, C. Autophagy Proteins in Viral Exocytosis and Anti-Viral Immune Responses. Viruses 2017, 9, 288. https://doi.org/10.3390/v9100288
Münz C. Autophagy Proteins in Viral Exocytosis and Anti-Viral Immune Responses. Viruses. 2017; 9(10):288. https://doi.org/10.3390/v9100288
Chicago/Turabian StyleMünz, Christian. 2017. "Autophagy Proteins in Viral Exocytosis and Anti-Viral Immune Responses" Viruses 9, no. 10: 288. https://doi.org/10.3390/v9100288
APA StyleMünz, C. (2017). Autophagy Proteins in Viral Exocytosis and Anti-Viral Immune Responses. Viruses, 9(10), 288. https://doi.org/10.3390/v9100288