
Gene Regulation and Quality Control in Murine Polyomavirus Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
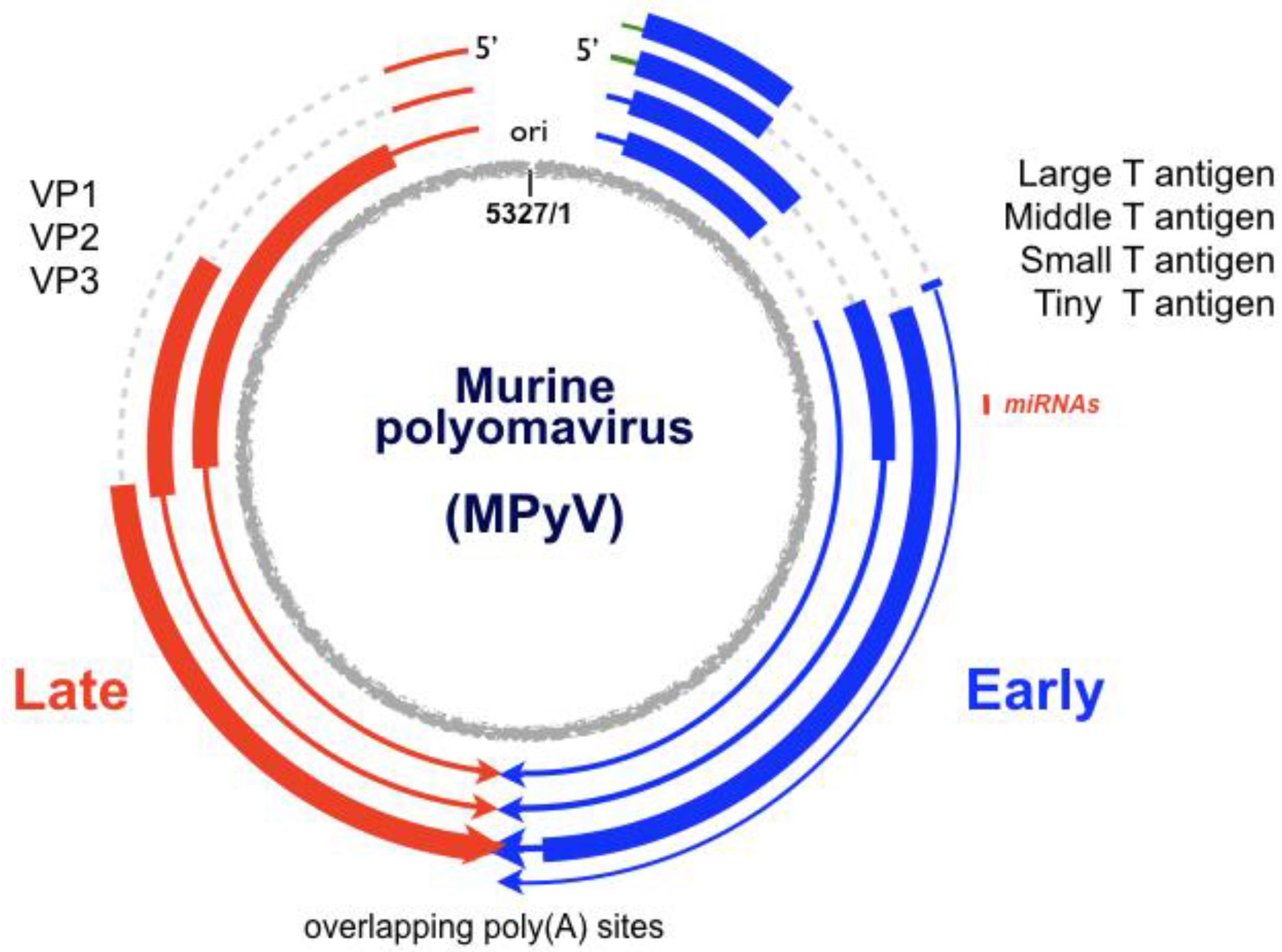
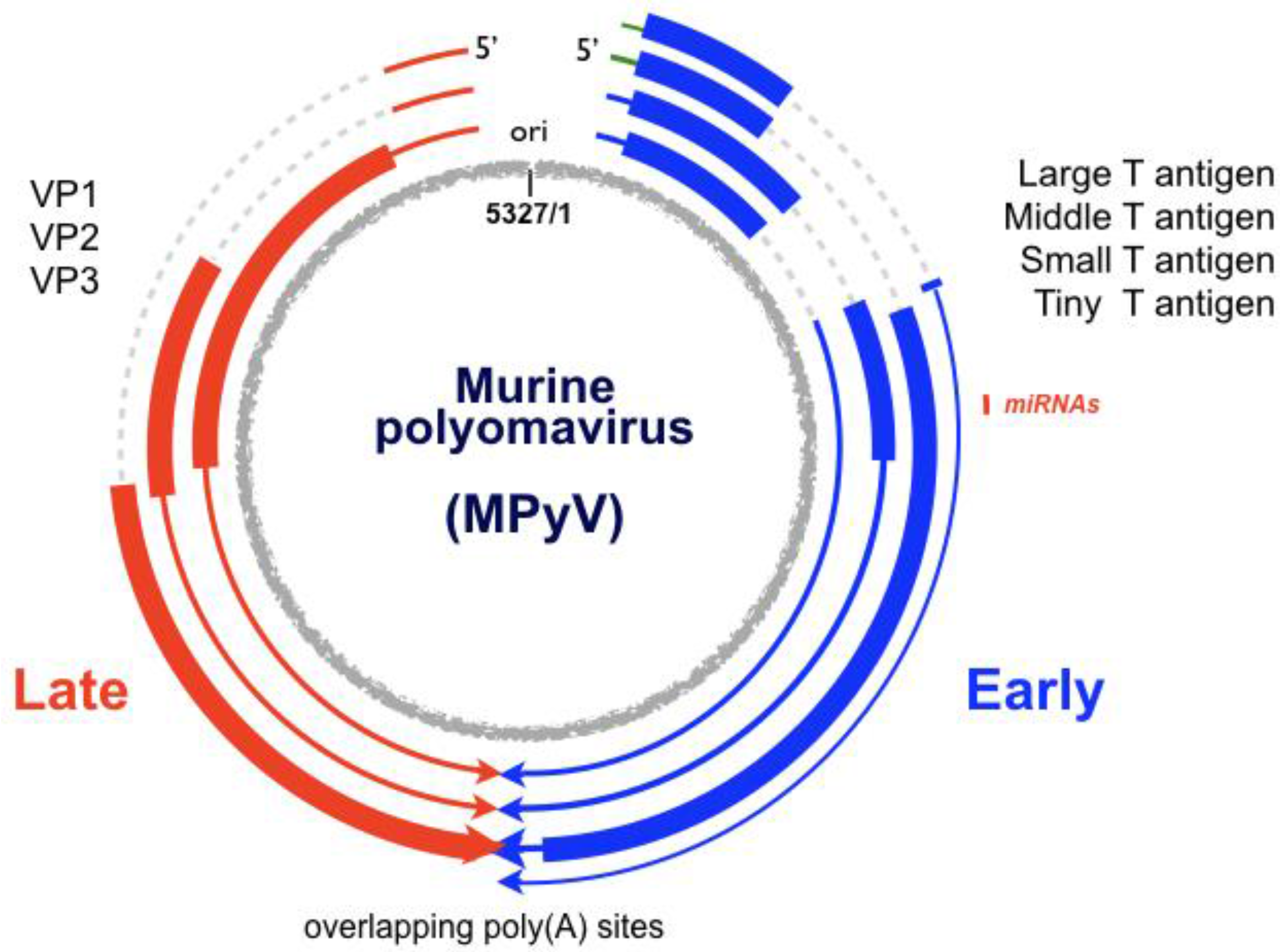
:1. The Virus
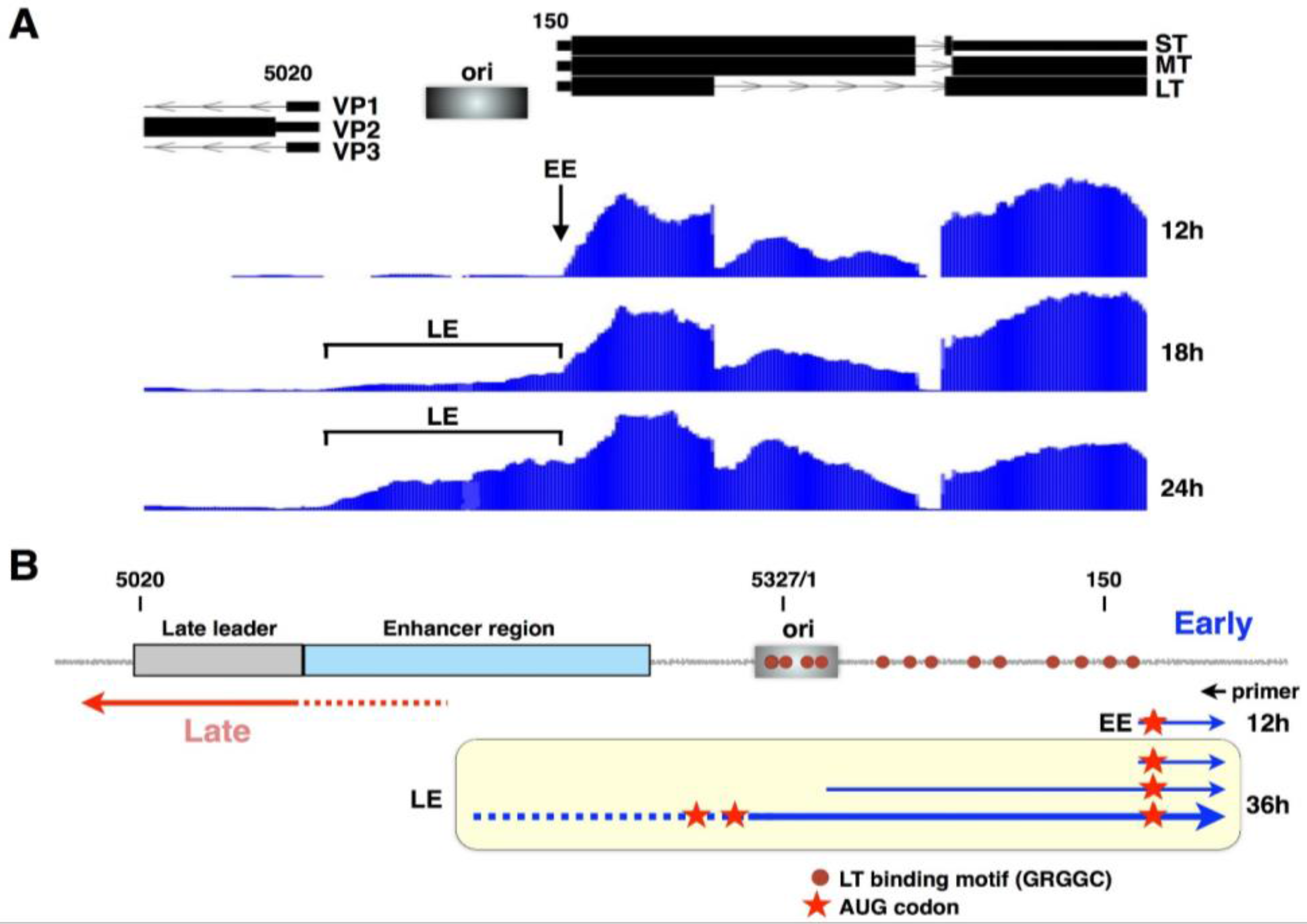
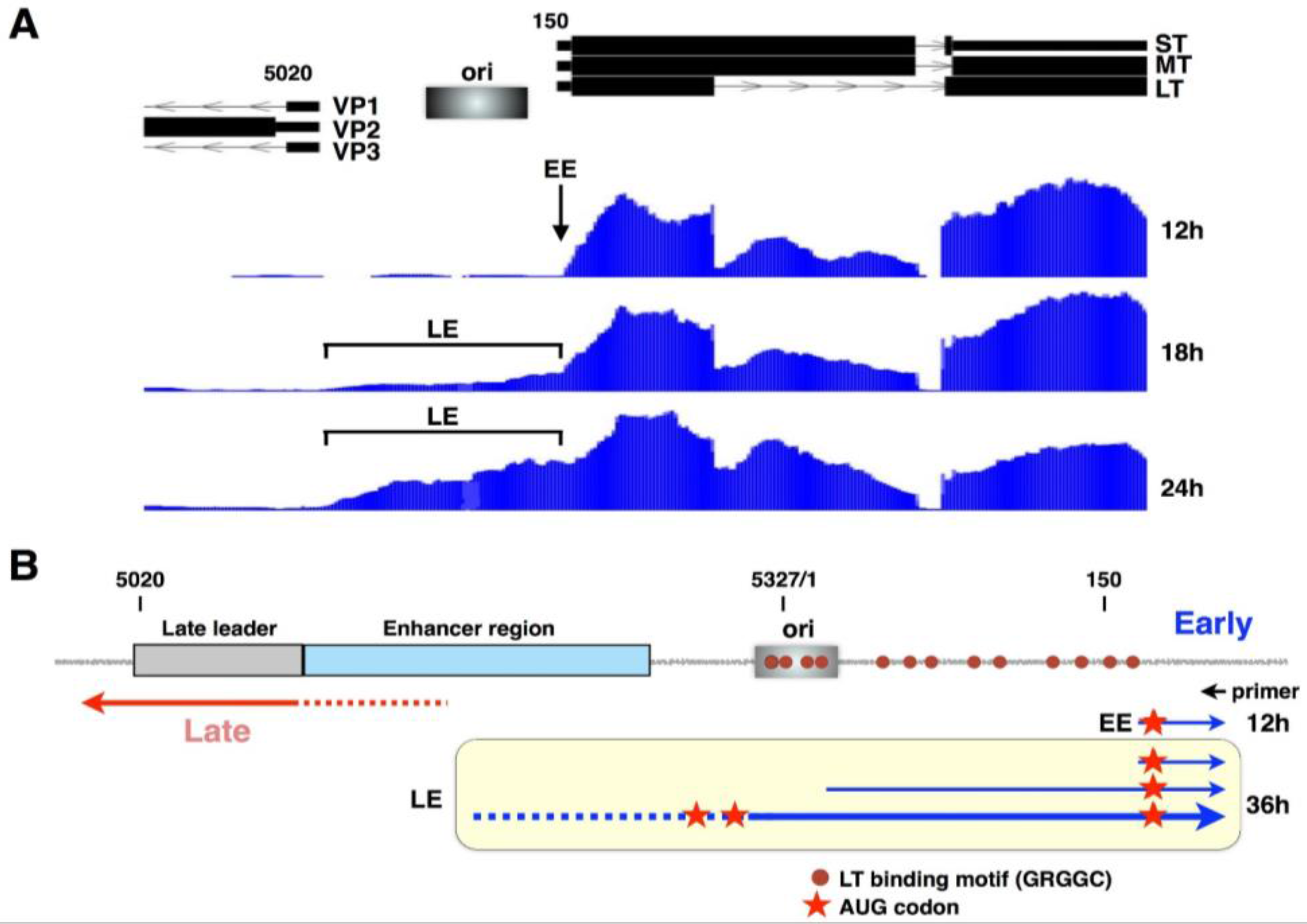
2. The Viral Early–Late Switch
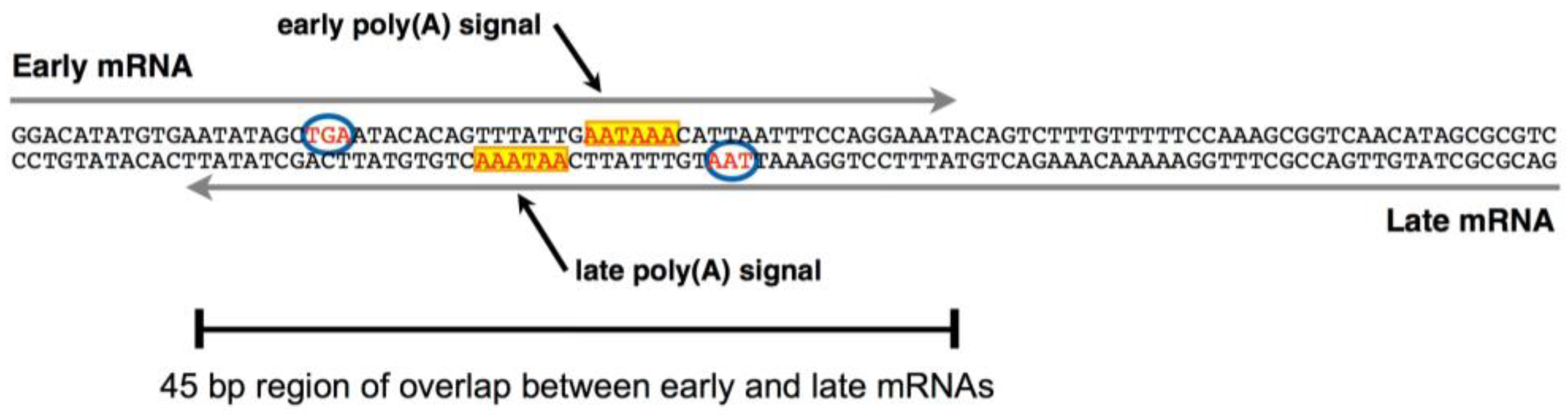
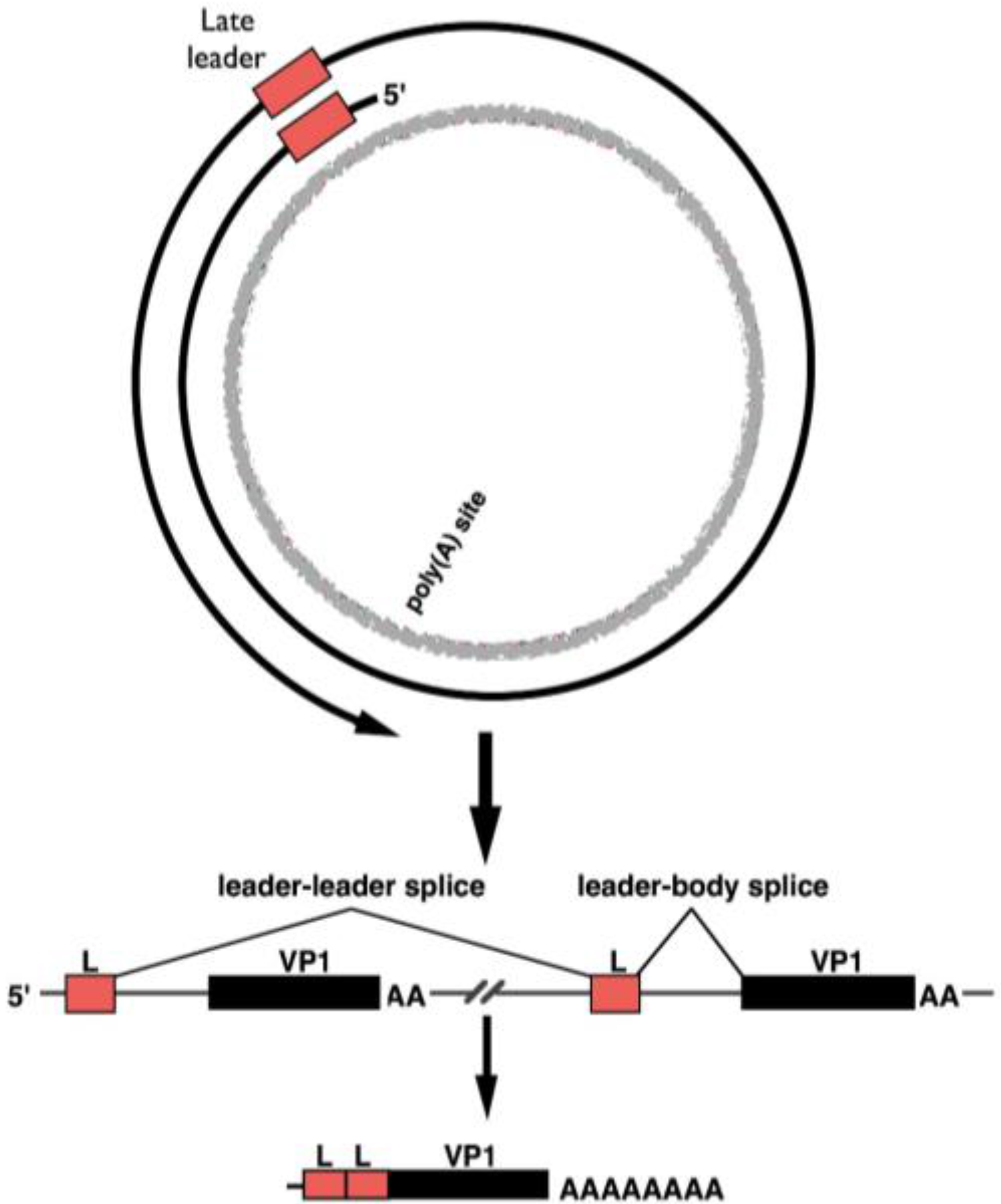
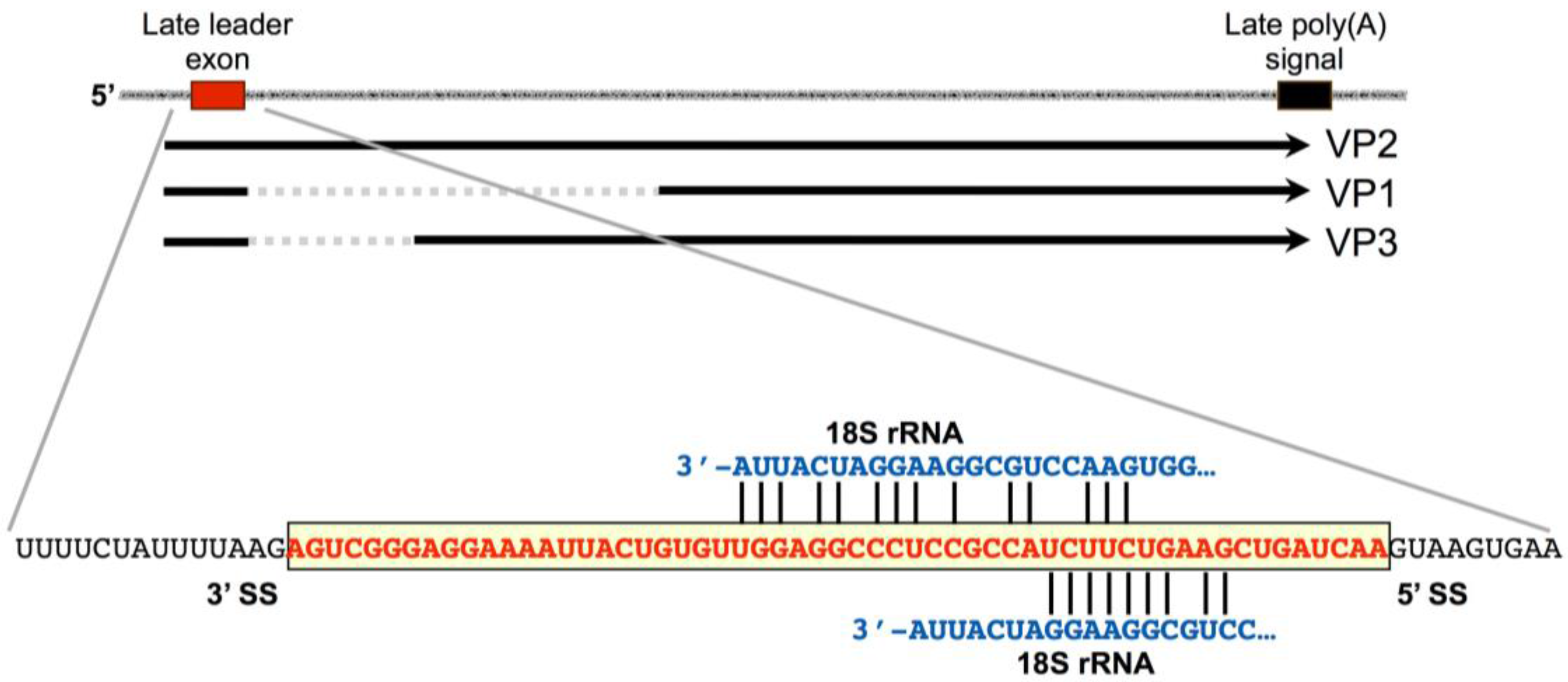
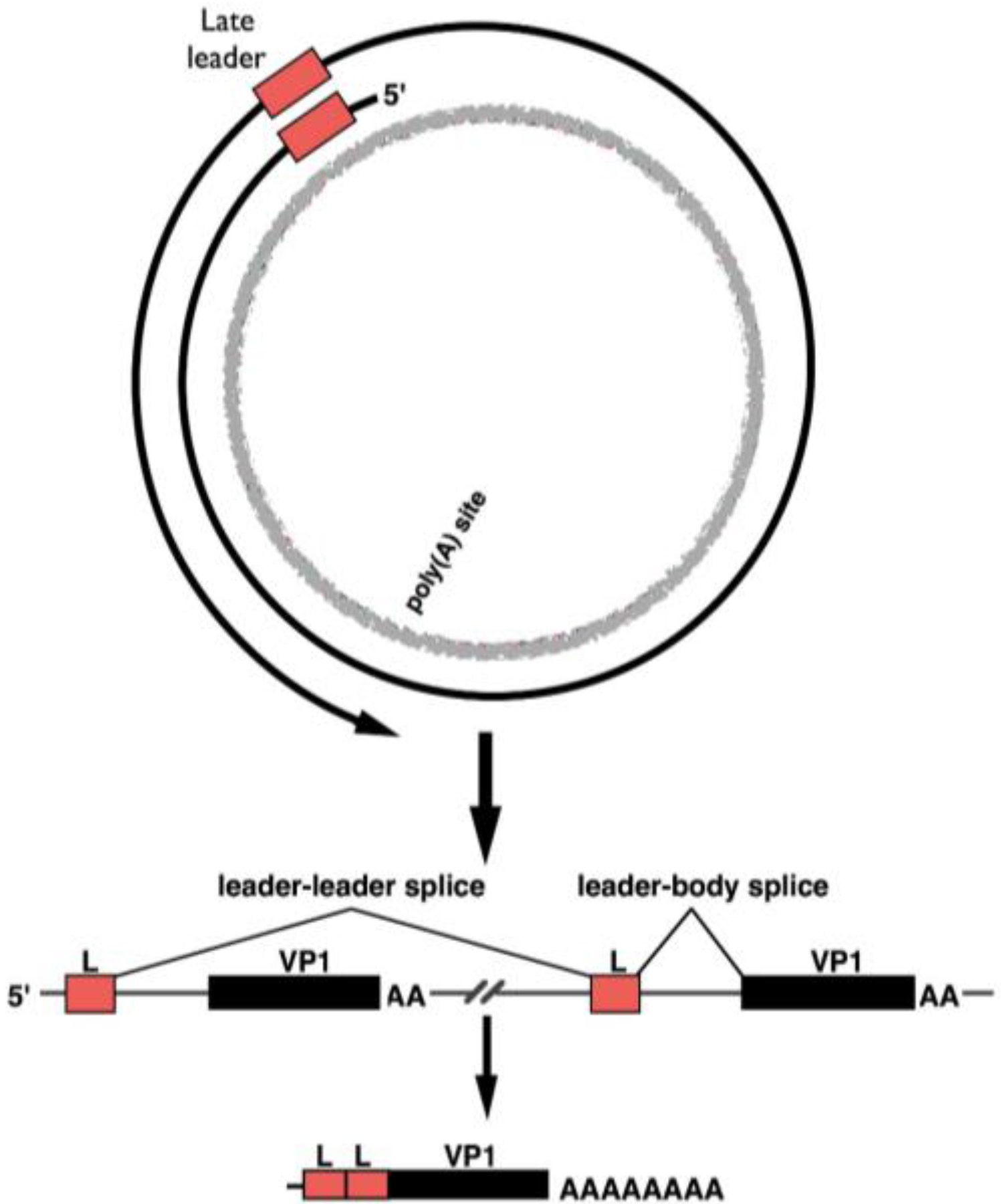
3. Late-Strand RNAs
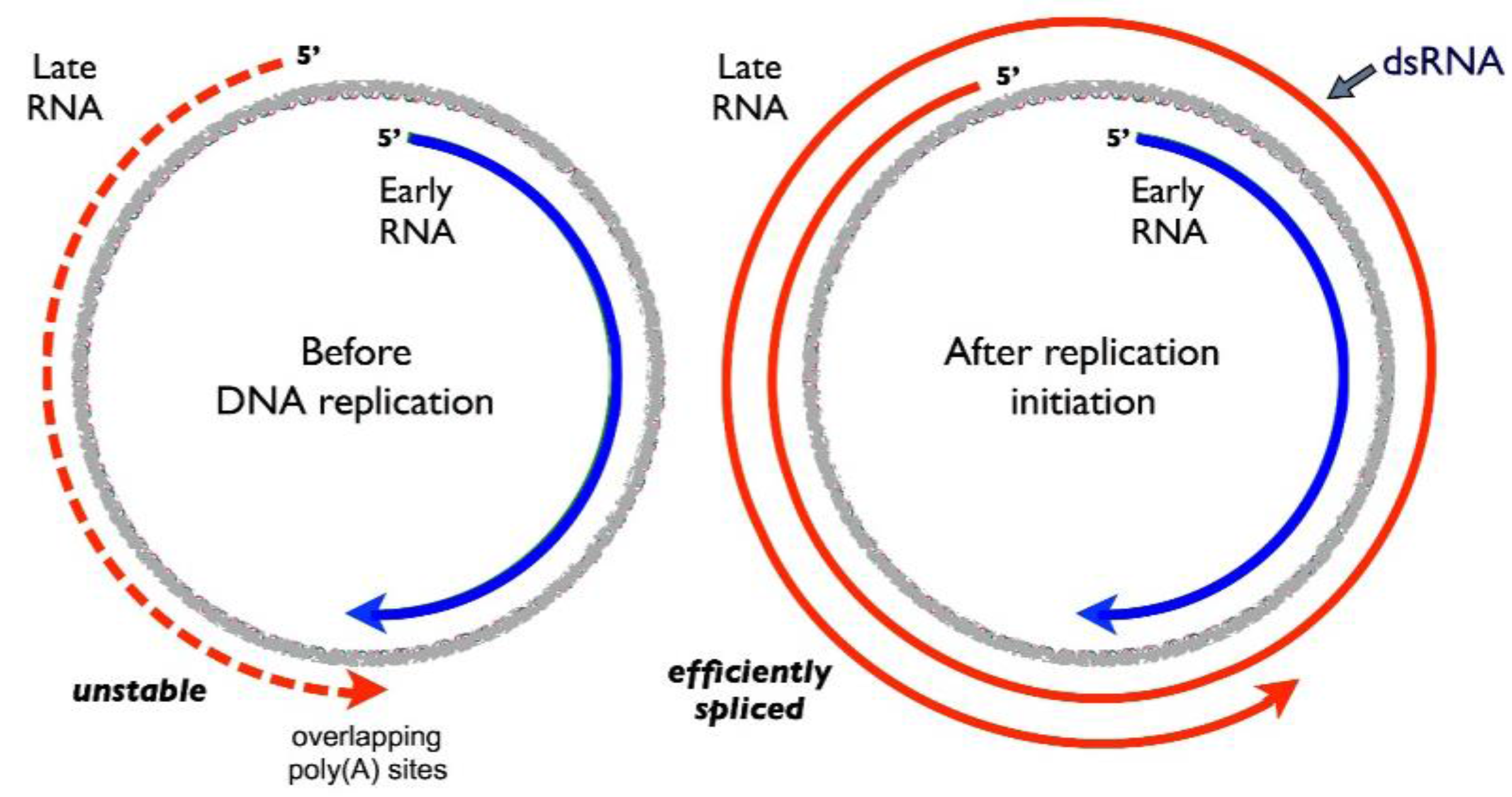
4. Activation of Late RNA Accumulation
5. The Role of dsRNA Formation and A-To-I Editing in MPyV Gene Regulation
6. Early-Strand RNAs
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kamen, R.; Lindstrom, D.M.; Shure, H.; Old, R.W. Virus-specific RNA in cells productively infected or transformed by polyoma virus. Cold Spring Harb. Symp. Quant. Biol. 1974, 39, 187–198. [Google Scholar] [CrossRef]
- Griffin, B.; Fried, M. Amplification of a specific region of the polyoma virus genome. Nature 1975, 256, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Kamen, R.; Favaloro, J.; Parker, J. Topography of the three late mRNA’s of polyoma virus which encode the virion proteins. J. Virol. 1980, 33, 637–651. [Google Scholar] [PubMed]
- Kamen, R.; Favaloro, J.; Parker, J.; Treisman, R.; Lania, L.; Fried, M.; Mellor, A. A comparison of polyomavirus transcription in productively infected mouse cells and transformed rodent cell lines. Cold Spring Harb. Symp. Quant. Biol. 1980, 44, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.; Robbins, A.; Nicklin, P. Location of the origin and terminus of replication in polyoma virus DNA. J. Gen. Virol. 1974, 25, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.I.; Yoo, W.; Mda, N.Y.; Folk, W.R. Tiny T antigen: An autonomous polyomavirus T antigen amino-terminal domain. J. Virol. 1997, 71, 6068–6074. [Google Scholar] [PubMed]
- Gaudray, P.; Tyndall, C.; Kamen, R.; Cuzin, F. The high affinity binding site on polyoma virus DNA for the viral large-T protein. Nucleic Acids Res. 1981, 9, 5697–5710. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, B.J.; Mueller, C.R.; Hassell, J.A. Polyomavirus large T antigen binds independently to multiple, unique regions on the viral genome. J. Virol. 1983, 47, 600–610. [Google Scholar] [PubMed]
- Cowie, A.; Kamen, R. Multiple binding sites for polyomavirus large T antigen within regulatory sequences of polyomavirus DNA. J. Virol. 1984, 52, 750–760. [Google Scholar] [PubMed]
- Dilworth, S.; Cowie, A.; Kamen, R.; Griffin, B. DNA binding activity of polyoma virus large tumor antigen. Proc. Natl. Acad. Sci. USA 1984, 81, 1941–1945. [Google Scholar] [CrossRef] [PubMed]
- Cogen, B. Virus-specific early RNA in 3T6 cells infected by a tsA mutant of polyoma virus. Virology 1978, 85, 222–230. [Google Scholar] [CrossRef]
- Farmerie, W.G.; Folk, W.R. Regulation of polyoma-virus transcription by large tumor antigen. Proc. Natl. Acad. Sci. USA 1984, 81, 6919–6923. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Carmichael, G.G. Polyoma virus early-late switch: Regulation of late RNA accumulation by DNA replication. Proc. Natl. Acad. Sci. USA 1993, 90, 8494–8498. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.B.; Roome, A.J.; Carmichael, G.G. Replication-dependent transactivation of the polyomavirus late promoter. J. Virol. 1990, 64, 992–1001. [Google Scholar] [PubMed]
- Cheng, J.; DeCaprio, J.A.; Fluck, M.M.; Schaffhausen, B.S. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin. Cancer Biol. 2009, 19, 218–228. [Google Scholar] [CrossRef] [PubMed]
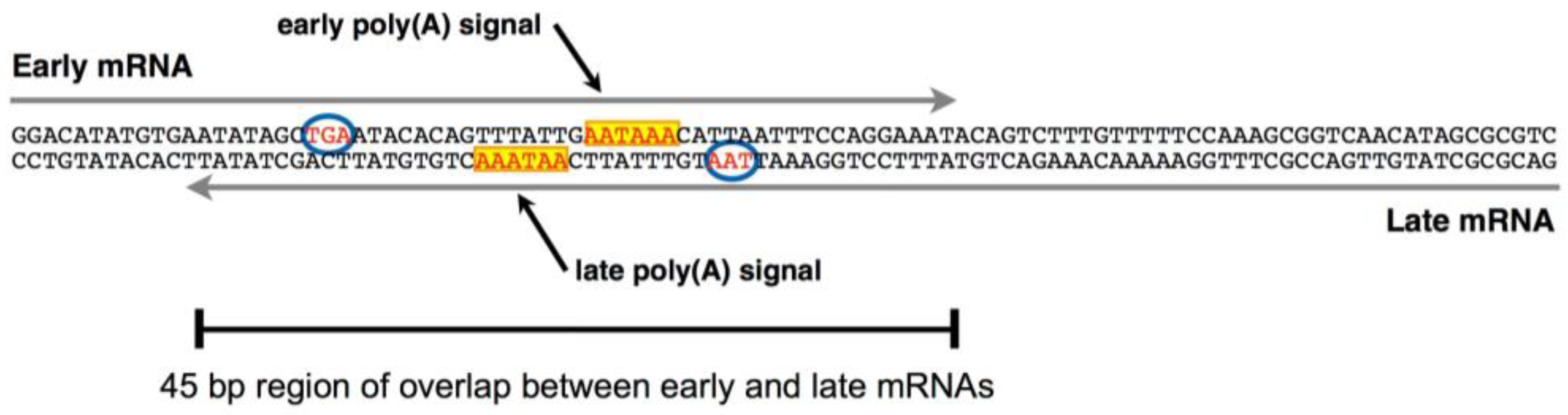
- Gu, R.; Zhang, Z.; DeCerbo, J.N.; Carmichael, G.G. Gene regulation by sense-antisense overlap of polyadenylation signals. RNA 2009, 15, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Beard, P.; Acheson, N.H.; Maxwell, I.H. Strand-specific transcription of polyoma virus DNA early in productive infection and in transformed cells. J. Virol. 1976, 17, 20–26. [Google Scholar]
- Piper, P. Polyoma virus transcription early during productive infection of mouse 3T6 cells. J. Mol. Biol. 1979, 131, 399–407. [Google Scholar] [CrossRef]
- Hyde-DeRuyscher, R.; Carmichael, G.G. Polyomavirus early-late switch is not regulated at the level of transcription initiation and is associated with changes in RNA processing. Proc. Natl. Acad. Sci. USA 1988, 85, 8993–8997. [Google Scholar] [CrossRef] [PubMed]
- Garren, S.B.; Kondaveeti, Y.; Duff, M.O.; Carmichael, G.G. Global Analysis of Mouse Polyomavirus Infection Reveals Dynamic Regulation of Viral and Host Gene Expression and Promiscuous Viral RNA Editing. PLoS Pathog. 2015, 11, e1005166. [Google Scholar] [CrossRef] [PubMed]
- Heiser, W.C.; Eckhart, W. Polyoma virus early and late mRNAs in productively infected mouse 3T6 cells. J. Virol. 1982, 44, 175–188. [Google Scholar] [PubMed]
- Kamen, R.; Jat, P.; Treisman, R.; Favaloro, J.; Folk, W.R. 5′ Termini of polyoma virus early region transcripts synthesizedin vivo by wild-type virus and viable deletion mutants. J. Mol. Biol. 1982, 159, 189–224. [Google Scholar] [CrossRef]
- Hyde-DeRuyscher, R.P.; Carmichael, G.G. Polyomavirus late pre-mRNA processing: DNA-replication-associated changes in leader exon multiplicity suggest a role for leader-to-leader splicing in the early-late switch. J. Virol. 1990, 64, 5823–5832. [Google Scholar] [PubMed]
- Liu, Z.; Batt, D.B.; Carmichael, G.G. Targeted nuclear antisense RNA mimics natural antisense-induced degradation of polyoma virus early RNA. Proc. Natl. Acad. Sci. USA 1994, 91, 4258–4262. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Sung, C.K.; Pack, C.D.; Grundhoff, A.; Lukacher, A.E.; Benjamin, T.L.; Ganem, D. Murine Polyomavirus encodes a microRNA that cleaves early RNA transcripts but is not essential for experimental infection. Virology 2009, 387, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.R.; Marlor, C.W.; Barrett, N.L.; Carmichael, G.G. Leader-to-leader splicing is required for efficient production and accumulation of polyomavirus late mRNAs. J. Virol. 1989, 63, 85–93. [Google Scholar] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Means to an end: Mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdiscip. Rev. RNA 2014, 5, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Acheson, N.; Buetti, E.; Scherrer, K.; Weil, R. Transcription of the polyoma virus genome: Synthesis and cleavage of giant late polyoma specific RNA. Proc. Natl. Acad. Sci. USA 1971, 68, 2231–2235. [Google Scholar] [CrossRef] [PubMed]
- Acheson, N. Transcription during productive infection with polyoma virus and SV40. Cell 1976, 8, 1–12. [Google Scholar] [CrossRef]
- Birg, F.; Favaloro, J.; Kamen, R. Analysis of polyoma viral nuclear RNA by miniblot hybridization. Proc. Natl. Acad. Sci. USA 1977, 74, 3138–3142. [Google Scholar] [CrossRef] [PubMed]
- Acheson, N.H. Polyoma giant RNAs contain tandem repeats of the nucleotide sequence of the entire viral genome. Proc. Natl. Acad. Sci. USA 1978, 75, 4754–4758. [Google Scholar] [CrossRef] [PubMed]
- Treisman, R. Characterization of polyoma late mRNA leader sequences by molecular cloning and DNA sequence analysis. Nucl. Acids Res. 1980, 8, 4867–4888. [Google Scholar] [CrossRef] [PubMed]
- Treisman, R.; Kamen, R. Structure of polyoma virus late nuclear RNA. J. Mol. Biol. 1981, 148, 273–301. [Google Scholar] [CrossRef]
- Luo, Y.; Carmichael, G.G. Splice site skipping in polyomavirus late pre-mRNA processing. J. Virol. 1991, 65, 6637–6644. [Google Scholar] [PubMed]
- Acheson, N. Kinetics and efficiency of polyadenylation of late polyomavirus nuclear RNA: Generation of oligomeric polyadenylated RNAs and their processing into mRNA. Mol. Cell. Biol. 1984, 4, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.K.; Yim, H.; Andrews, E.; Benjamin, T.L. A mouse polyomavirus-encoded microRNA targets the cellular apoptosis pathway through Smad2 inhibition. Virology 2014, 468–470, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Carmichael, G.G. Splice site choice in a complex transcription unit containing multiple inefficient polyadenylation signals. Mol. Cell. Biol. 1991, 11, 5291–5300. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Carmichael, G.G. A suboptimal 5′ splice site is a cis-acting determinant of nuclear export of polyomavirus late mRNAs. Mol. Cell. Biol. 1996, 16, 6046–6054. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Carmichael, G.G. Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc. Natl. Acad. Sci. USA 1997, 94, 3542–3547. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Zhang, Z.; Carmichael, G.G. How a small DNA virus uses dsRNA but not RNAi to regulate its life cycle. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 293–299. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Samuel, C.E. Host response to polyomavirus infection is modulated by RNA adenosine deaminase ADAR1 but not by ADAR2. J. Virol. 2011, 85, 8338–8347. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.G.; Pellegrini, S.; Cowie, A.; Basilico, C. Regulation of polyomavirus late promoter activity by viral early proteins. J. Virol. 1986, 60, 275–285. [Google Scholar] [PubMed]
- Zhang, Z.; Carmichael, G.G. The fate of dsRNA in the nucleus. A p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 2001, 106, 465–475. [Google Scholar] [CrossRef]
- Chen, L.L.; Carmichael, G.G. Gene regulation by SINES and inosines: Biological consequences of A-to-I editing of Alu element inverted repeats. Cell Cycle 2008, 7, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: Functional role of a nuclear noncoding RNA. Mol. Cell 2009, 35, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Fenton, R.G.; Basilico, C. Changes in the topography of early region transcription during polyomavirus lytic infection. Proc. Natl. Acad. Sci. USA 1982, 79, 7142–7146. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.K.; Lebowitz, P. Simian virus early mRNA’s contain multiple 5′ termini upstream and downstream from a Hogness-Goldberg sequence; a shift in 5′ termini during the lytic cycle mediated by large T antigen. J. Virol. 1981, 40, 224–240. [Google Scholar] [PubMed]
- Khalili, K.; Feigenbaum, L.; Khoury, G. Evidence for a shift in 5′-termini of early viral RNA during the lytic cycle of JC virus. Virology 1987, 158, 469–472. [Google Scholar] [CrossRef]
- Carbone, M.; Hauser, J.; Carty, M.P.; Rundell, K.; Dixon, K.; Levine, A.S. Simian virus-40 (SV40) small T-antigen inhibits SV40 DNA replication in vitro. J. Virol. 1992, 66, 1804–1808. [Google Scholar] [PubMed]
- Theiss, J.M.; Gunther, T.; Alawi, M.; Neumann, F.; Tessmer, U.; Fischer, N.; Grundhoff, A. A Comprehensive Analysis of Replicating Merkel Cell Polyomavirus Genomes Delineates the Viral Transcription Program and Suggests a Role for mcv-miR-M1 in Episomal Persistence. PLoS Pathog. 2015, 11, e1004974. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmichael, G.G. Gene Regulation and Quality Control in Murine Polyomavirus Infection. Viruses 2016, 8, 284. https://doi.org/10.3390/v8100284
Carmichael GG. Gene Regulation and Quality Control in Murine Polyomavirus Infection. Viruses. 2016; 8(10):284. https://doi.org/10.3390/v8100284
Chicago/Turabian StyleCarmichael, Gordon G. 2016. "Gene Regulation and Quality Control in Murine Polyomavirus Infection" Viruses 8, no. 10: 284. https://doi.org/10.3390/v8100284
APA StyleCarmichael, G. G. (2016). Gene Regulation and Quality Control in Murine Polyomavirus Infection. Viruses, 8(10), 284. https://doi.org/10.3390/v8100284