
Innate Immunity Evasion by Enteroviruses: Insights into Virus-Host Interaction

Abstract
:1. Enterovirus
1.1. Pathogenesis

{kind=link}
| Enterovirus Species | Type |
|---|---|
| Human enterovirus A | Human coxsackievirus A2–8, 10, 12, 14, 16. |
| Human enterovirus 71, 76, 89–92, 114, 119–121. | |
| Human enterovirus B | Human coxsackievirus A-9, B1-6. |
| Human echovirus 1–9, 11–21, 24–27, 29–33. | |
| Human enterovirus 69, 73–75, 77–88, 93, 97–101, 106, 107, 110–113. | |
| Human enterovirus C | Human coxsackievirus A1, 11, 13, 17, 19–22, 24. |
| Human poliovirus 1–3. | |
| Human enterovirus 95, 96, 99, 102, 104, 105, 109, 116–118. | |
| Human enterovirus D | Human enterovirus 68, 70, 94, 111, 120. |
| Rhinovirus A | Human rhinovirus A1, 2, 7–13, 15, 16, 18–25, 28–34, 36, 38–41, 43, 45–47, 49–51, 53–68, 71, 73–78, 80–82, 85, 88–90, 94, 96, 100–109. |
| Rhinovirus B | Human rhinovirus B3-6, 14, 17, 26, 27, 35, 37, 42, 48, 52, 69, 70, 72, 79, 83, 84, 86, 91–93, 97, 99, 100–106. |
| Rhinovirus C | Human rhinovirus C1-55. |
1.2. Enteroviruses (EV) Genome and Viral Proteins
2. Innate Immunity
2.1. Innate Immunity Signaling Pathways
2.2. Innate Detection of Enterovirus
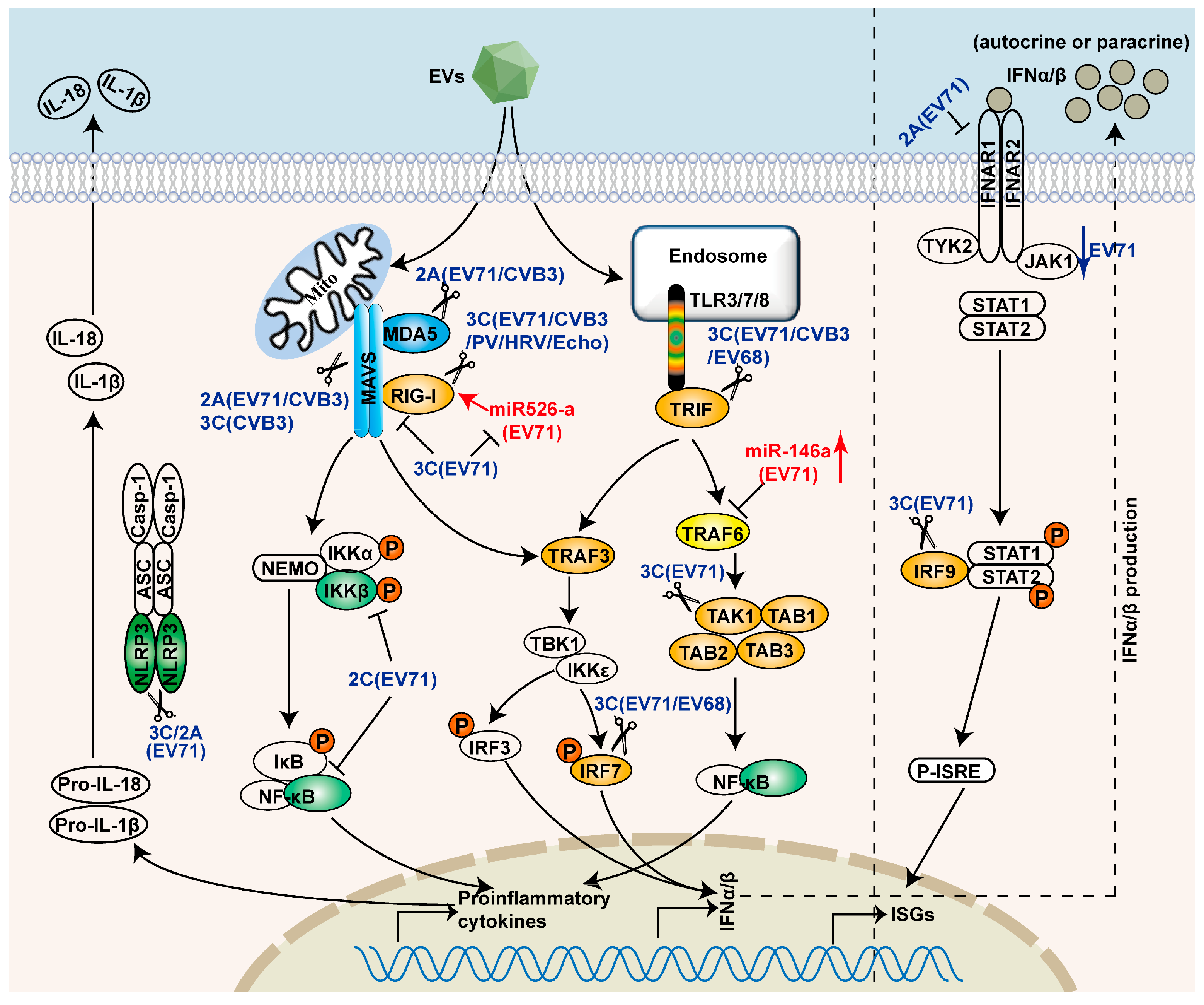
3. Evasion of Innate Immunity by EVs
3.1. Shutoff of Host Protein Synthesis
| Targets | Viral Proteins | Antagonizing Approach | References |
|---|---|---|---|
| RIG-I | EV-A71 3C | Impeding ofinteraction between RIG-I and MAVS | [26] |
| CV-B3 3C | Cleavage | [56] | |
| PV 3C | Cleavage | [56,70] | |
| Rhinovirus 16 3C | Cleavage | [70] | |
| Echovirus 3C | Cleavage | [70] | |
| MDA-5 | EV-A71 induced caspases | Cleavage | [57] |
| PV induced caspases | Degradation in proteasome- and caspase-dependent manner | [71] | |
| EV-A71 2A | Cleavage | [56] | |
| CV-B3 2A | Cleavage | [56] | |
| PV 2A | Cleavage | [56] | |
| MAVS | EV-A71 2A | Cleavage | [28,56] |
| PV2A | Cleavage | [56] | |
| PV induced caspases | Cleavage | [72] | |
| CV-B3 3C | Cleavage | [73] | |
| CV-B3 2A | Cleavage | [56] | |
| HRV1a 2A, 3C and caspases | Cleavage | [74] | |
| TRIF | EV-A71 3C | Cleavage | [75] |
| EV-D68 3C | Cleavage | [76] | |
| CV-B3 3C | Cleavage | [73] | |
| IRF7 | EV-A71 3C | Cleavage | [77] |
| EV-D68 3C | Cleavage | [78] | |
| IRF9 | EV-A71 3C | Cleavage | [79] |
| IKKβ | EV-A71 2C | Inhibition of phosphorylation | [80] |
| P65 | EV-A71 2C | Inhibition of interaction between P65 and P50 | [81] |
| TAK1 Complex | EV-A71 3C | Cleavage of TAK1/TAB1/TAB2/TAB3 | [82] |
| IFANR1 | EV-A71 2A | Down-regulation | [83] |
| G3BP1 | CV-B3 3C | Cleavage | [84] |
| miR-146a | EV-A71 | Upregulation of miR-146a to inhibit IRAK1- and TRAF6-mediated IFNβ production | [85] |
| miR-526a | EV-A71 3C | Downregulation | [86] |

3.2. Interference with PRRs Recognition
3.3. Interference with Adaptor Molecules and Downstream Effectors in Innate Immune Signaling Pathways
3.4. Interference with IFN-Mediated Signaling
3.5. Interference of Other Signaling Pathways
4. Roles of Innate Immunity Evasion in EV Pathogenesis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- The Pirbright Institute, UK. Available online: http://www.picornaviridae.com/enterovirus/enterovirus.htm (accessed on 10 October 2015).
- Racaniello, V.R. Picornarviridae: The Viruses and Their Replication. In Fielda Virology; Howley, P.M., Knipe, D.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Tebruegge, M.; Curtis, N. Enterovirus infections in neonates. Semin. Fetal Neonatal Med. 2009, 14, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Precechtelova, J.; Borsanyiova, M.; Sarmirova, S.; Bopegamage, S. Type I diabetes mellitus: Genetic factors and presumptive enteroviral etiology or protection. J. Pathog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Whitton, J.L.; Cornell, C.T.; Feuer, R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 2005, 3, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Kurten, M.; Kennedy, J.L. Rhinovirus and asthma: A storied history of incompatibility. Curr. Allergy Asthma Rep. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Stone, C.A., Jr.; Miller, E.K. Understanding the association of human rhinovirus with asthma. Clin. Vaccine Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Glanville, N.; Johnston, S.L. Challenges in developing a cross-serotype rhinovirus vaccine. Curr. Opin. Virol. 2015, 11, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Naccache, S.N.; Messacar, K.; Clayton, A.; Yu, G.; Somasekar, S.; Federman, S.; Stryke, D.; Anderson, C.; Yagi, S.; et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012–2014): A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 671–682. [Google Scholar] [CrossRef]
- Khan, F. Enterovirus D68: Acute respiratory illness and the 2014 outbreak. Emerg. Med. Clin. North Am. 2015, 33, e19–e32. [Google Scholar] [CrossRef] [PubMed]
- Waghmare, A.; Pergam, S.A.; Jerome, K.R.; Englund, J.A.; Boeckh, M.; Kuypers, J. Clinical disease due to enterovirus D68 in adult hematologic malignancy patients and hematopoietic cell transplant recipients. Blood 2015, 125, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Schnurr, D.; Flemister, M.R.; Lovchik, J.C.; Peters, H.; Sessions, W.; Kirk, C.; Chatterjee, N.; Fuller, S.; et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J. Gen. Virol. 2004, 85, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Puenpa, J.; Mauleekoonphairoj, J.; Linsuwanon, P.; Suwannakarn, K.; Chieochansin, T.; Korkong, S.; Theamboonlers, A.; Poovorawan, Y. Prevalence and characterization of enterovirus infections among pediatric patients with hand foot mouth disease, herpangina and influenza like illness in Thailand, 2012. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Du, J.; Xue, Y.; Su, H.; Yang, F.; Jin, Q. Epidemics and frequent recombination within species in outbreaks of human enterovirusB-associated hand, foot and mouth disease in Shandong China in 2010 and 2011. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Chan, K.P.; Goh, K.T.; Chong, C.Y.; Teo, E.S.; Lau, G.; Ling, A.E. Epidemic hand, foot and mouth disease caused by human enterovirus 71, Singapore. Emerg. Infect. Dis. 2003, 9, 78–85. [Google Scholar] [CrossRef] [PubMed]
- McMinn, P.; Stratov, I.; Nagarajan, L.; Davis, S. Neurological manifestations of enterovirus 71 infection in children during an outbreak of hand, foot, and mouth disease in Western Australia. Clin. Infect. Dis. 2001, 32, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y.; et al. Hand, foot, and mouth disease in China, 2008–2012: An epidemiological study. Lancet Infect. Dis. 2014, 14, 308–318. [Google Scholar] [CrossRef]
- Wang, S.M.; Lei, H.Y.; Yu, C.K.; Wang, J.R.; Su, I.J.; Liu, C.C. Acute chemokine response in the blood and cerebrospinal fluid of children with enterovirus 71-associated brainstem encephalitis. J. Infect. Dis. 2008, 198, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- WHO. A Guide to Clinical Management and Public Health Response for Hand, Foot and Mouth Disease (HFMD); WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Yang, F.; Ren, L.; Xiong, Z.; Li, J.; Xiao, Y.; Zhao, R.; He, Y.; Bu, G.; Zhou, S.; Wang, J.; et al. Enterovirus 71 outbreak in the People’s Republic of China in 2008. J. Clin. Microbiol. 2009, 47, 2351–2352. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Notes from the field: Severe hand, foot, and mouth disease associated with coxsackievirus A6-Alabama, Connecticut, California, and Nevada, November 2011–February 2012. MMWR Morb. Mortal. Weekly Rep. 2012, 61, 213–214. [Google Scholar]
- Li, J.L.; Yuan, J.; Yang, F.; Wu, Z.Q.; Hu, Y.F.; Xue, Y.; Zhou, B.P.; Jin, Q. Epidemic characteristics of hand, foot, and mouth disease in southern China, 2013: Coxsackievirus A6 has emerged as the predominant causative agent. J. Infect. 2014, 69, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Osterback, R.; Vuorinen, T.; Linna, M.; Susi, P.; Hyypia, T.; Waris, M. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg. Infect. Dis. 2009, 15, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Krausslich, H.G.; Nicklin, M.J.; Toyoda, H.; Etchison, D.; Wimmer, E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 1987, 61, 2711–2718. [Google Scholar] [PubMed]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Hsu, T.A.; Chen, T.C.; Chang, S.C.; Lee, J.C.; Chen, C.C.; Stollar, V.; Shih, S.R. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 2002, 293, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Weng, K.F.; Li, M.L.; Hung, C.T.; Shih, S.R. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Devasthanam, A.S. Mechanisms underlying the inhibition of interferon signaling by viruses. Virulence 2014, 5, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rich, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Mak, T.W.; Sen, G.; Li, X. Toll-like receptor 3-mediated activation of NF-κB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-β. Proc. Natl. Acad. Sci. USA 2004, 101, 3533–3538. [Google Scholar] [CrossRef] [PubMed]
- Muroi, M.; Tanamoto, K. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-κB. J. Leukoc. Biol. 2008, 83, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A conserved histidinein the RNA sensor RIG-I controls immune tolerance to N1-2’O-methylated self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Ruegg, A.; Werner, S.; Beer, H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Bozym, R.; Morosky, S.A.; Hanna, S.L.; Mukherjee, A.; Tudor, M.; Kim, K.S.; Cherry, S. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 2011, 9, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Asher, D.R.; Chan, M.; Kurt-Jones, E.A.; Finberg, R.W. Cutting Edge: Antibody-mediated TLR7-dependent recognition of viral RNA. J. Immunol. 2007, 178, 3363–3367. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.J.; Lavallee, D.J.; Shanina, I.; Horwitz, M.S. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS ONE 2009, 4, e4127. [Google Scholar] [CrossRef] [PubMed]
- Abston, E.D.; Coronado, M.J.; Bucek, A.; Onyimba, J.A.; Brandt, J.E.; Frisancho, J.A.; Kim, E.; Bedja, D.; Sung, Y.K.; Radtke, A.J.; et al. TLR3 deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirusB3 infection: Role for IL-4. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R267–R277. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Osawa, T.; Ogami, K.; Ouyang, X.; Sakaguchi, S.; Koshiba, R.; Yanai, H.; Seko, Y.; Shitara, H.; Bishop, K.; et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20446–20451. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Fujii, K.; Nagata, N.; Takeuchi, O.; Akira, S.; Oshiumi, H.; Matsumoto, M.; Seya, T.; Koike, S. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J. Virol. 2012, 86, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Chou, A.H.; Lin, S.I.; Chen, I.H.; Lien, S.P.; Liu, C.C.; Chong, P.; Liu, S.J. Toll-like receptor 9-mediated protection of enterovirus 71 infection in mice is due to the release of danger-associated molecular patterns. J. Virol. 2014, 88, 11658–11670. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.L.; Kao, L.T.; Lin, S.J.; Wang, R.Y.; Shih, S.R. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, B.; Xiong, S. Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1438–H1447. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lei, X.; Xiao, X.; Yang, C.; Lu, W.; Huang, Z.; Leng, Q.; Jin, Q.; He, B.; Meng, G.; et al. Reciprocal regulation between enterovirus71 and the NLRP3 inflammasome. Cell Rep. 2015, 12, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E.; Grubman, M.J.; Ehrenfeld, E. Relationship of p220 cleavage during picornavirus infection to 2A proteinase sequencing. J. Virol. 1988, 62, 4216–4223. [Google Scholar] [PubMed]
- Joachims, M.; van Breugel, P.C.; Lloyd, R.E. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 1999, 73, 718–727. [Google Scholar] [PubMed]
- Kerekatte, V.; Keiper, B.D.; Badorff, C.; Cai, A.; Knowlton, K.U.; Rhoads, R.E. Cleavage of Poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: Another mechanism for host protein synthesis shutoff? J. Virol. 1999, 73, 709–717. [Google Scholar] [PubMed]
- Kuyumcu-Martinez, N.M.; van Eden, M.E.; Younan, P.; Lloyd, R.E. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: A novel mechanism for host translation shutoff. Mol. Cell Biol. 2004, 24, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.E.; Lieberman, P.M.; Berk, A.J.; Dasgupta, A. Direct cleavage of human TATA-binding protein by poliovirus protease 3C in vivo and in vitro. Mol. Cell Biol. 1993, 13, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Weidman, M.K.; Yalamanchili, P.; Ng, B.; Tsai, W.; Dasgupta, A. Poliovirus 3C protease-mediated degradation of transcriptional activator p53 requires a cellular activity. Virology 2001, 291, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, P.; Harris, K.; Wimmer, E.; Dasgupta, A. Inhibition of basal transcription by poliovirus: A virus-Encodedprotease (3Cpro) inhibits formation of TBP-TATA box complex in vitro. J. Virol. 1996, 70, 2922–2929. [Google Scholar] [PubMed]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 391, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Meylan, E.; Curran, J.; Tschopp, J. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 2008, 15, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Drahos, J.; Racaniello, V.R. Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 2009, 83, 11581–11587. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Li, L.; Lei, X.; Zhou, H.; Zhou, Z.; He, B.; Wang, J. Enterovirus 68 3C protease cleaves TRIF to attenuate antiviral responses mediated by Toll-like receptor 3. J. Virol. 2014, 88, 6650–6659. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. The 3C protease of enterovirusD68 inhibits the cellular defense mediated by interferon regulatory factor 7. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Wang, H.C.; Shih, S.R.; Teng, I.F.; Tseng, C.P.; Hsu, J.T. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 2011, 203, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, H.; Zhang, Z.; Meng, J.; Mao, D.; Bai, B.; Lu, B.; Mao, P.; Hu, Q.; Wang, H. Enterovirus 71 2C protein inhibits TNF-α-mediated activation of NF-κB by suppressing IκB kinase β phosphorylation. J. Immunol. 2011, 187, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yin, P.; Yang, X.; Zhang, L.; Jin, Q.; Zhu, G. Enterovirus 71 2C Protein Inhibits NF-κB Activation by Binding to RelA(p65). Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Han, N.; Xiao, X.; Jin, Q.; He, B.; Wang, J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J. Virol. 2014, 88, 9830–9841. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yi, L.; Zhao, J.; Yu, J.; Chen, Y.; Lin, M.C.; Kung, H.F.; He, M.L. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 2012, 86, 3767–3776. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Ng, C.S.; Zhang, J.; Shi, J.; Wong, J.; Piesik, P.; Han, L.; Chu, F.; Jagdeo, J.; Jan, E.; et al. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS ONE 2013, 8, e79546. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Yu, I.S.; Lu, L.F.; Rudensky, A.; Chen, H.Y.; Tsai, C.W.; Chang, Y.L.; Wu, C.T.; Chang, L.Y.; Shih, S.R.; et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, X.; Zheng, Z.; Zhang, Z.; Wei, C.; Guan, K.; Hou, L.; Zhang, B.; Zhu, L.; Cao, Y.; et al. Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J. Virol. 2014, 88, 11356–11368. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, C.; Guo, N.; Zhu, K.; Luo, K.; Zhang, N.; Zhao, H.; Cui, Y.; Chen, L.; Wang, H.; et al. Type I interferons triggered through the Toll-like receptor 3-TRIF pathway control coxsackievirusA16 infection in young mice. J. Virol. 2015, 89, 10860–10867. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Yang, J.; Luo, K.; Yang, C.; Zhang, N.; Xu, R.; Chen, J.; Jin, M.; Xu, B.; Guo, N.; et al. TLR3 signaling in macrophages is indispensable for the protective immunity of invariant natural killer T cells against enterovirus 71 infection. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.P.; Wang, Y.F.; Wang, J.R.; Huang, S.W.; Yu, C.K. Enterovirus 71 blocks selectively type I interferon production through the 3C viral protein in mice. J. Med. Virol. 2012, 84, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Zhao, X.; Yu, R.; Zhang, X.; Wu, S.; Liu, J.; Chi, X.; Song, X.; Fu, L.; et al. Enterovirus 71 inhibits cellular type I interferon signaling by downregulating JAK1 protein expression. Viral Immunol. 2014, 27, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.D.; Semler, B.L. Poliovirus infection induces the co-localization of cellular protein SRp20 with TIA-1, a cytoplasmic stress granule protein. Virus Res. 2013, 176, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Lloyd, R.E. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Guo, X.; Qi, Y.; Qi, X.; Ge, Y.; Shi, Z.; Wu, T.; Shan, J.; Shan, Y.; Zhu, Z.; et al. Identification of microRNAs involved in the host response to enterovirus 71 infection by a deep sequencing approach. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Qi, Y.; Li, H.; Ge, Y.; Zhao, K.; Qi, X.; Guo, X.; Shi, Z.; Zhou, M.; Zhu, B.; et al. Serum microRNA expression profile distinguishes enterovirus 71 and coxsackievirus 16 infections in patients with hand-foot-and-mouth disease. PLoS ONE 2011, 6, e27071. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shen, L.; Chen, J.; Xu, H.; Mao, L. The role of microRNAs in enteroviral infections. Braz. J. Infect. Dis. 2015, 19, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Ida-Hosonuma, M.; Iwasaki, T.; Yoshikawa, T.; Nagata, N.; Sato, Y.; Sata, T.; Yoneyama, M.; Fujita, T.; Taya, C.; Yonekawa, H.; et al. The α/β interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 2005, 79, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Lee, Y.P.; Wang, Y.F.; Lei, H.Y.; Liu, C.C.; Wang, S.M.; Su, I.J.; Wang, J.R.; Yeh, T.M.; Chen, S.H.; et al. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 2005, 86, 3263–3269. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.; Xiao, X.; Wang, J. Innate Immunity Evasion by Enteroviruses: Insights into Virus-Host Interaction. Viruses 2016, 8, 22. https://doi.org/10.3390/v8010022
Lei X, Xiao X, Wang J. Innate Immunity Evasion by Enteroviruses: Insights into Virus-Host Interaction. Viruses. 2016; 8(1):22. https://doi.org/10.3390/v8010022
Chicago/Turabian StyleLei, Xiaobo, Xia Xiao, and Jianwei Wang. 2016. "Innate Immunity Evasion by Enteroviruses: Insights into Virus-Host Interaction" Viruses 8, no. 1: 22. https://doi.org/10.3390/v8010022
APA StyleLei, X., Xiao, X., & Wang, J. (2016). Innate Immunity Evasion by Enteroviruses: Insights into Virus-Host Interaction. Viruses, 8(1), 22. https://doi.org/10.3390/v8010022