
Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC


Abstract
:1. Introduction

{kind=link}
{kind=link}
| Species | Types of Viruses Detected Occasionally in Respiratory Samples | Types of Viruses Detected Predominantly or Exclusively in Respiratory Samples |
|---|---|---|
| EV-A | CV-A10, CV-A16, EV-A71 | – |
| EV-B | CV-A9, CV-B1, CV-B2, CV-B3, CV-B4, CV-B5, CV-B6, E-1, E-2, E-3, E-4, E-5, E-6, E-7, E-9, E-11, E-12, E-13, E-14, E-15, E-16, E-17, E-18, E-19, E-20, E-21, E-25, E-29, E-30 | – |
| EV-C | CV-A24, PV-3 | EV-C104 [6], EV-C105 [7], EV-C109 [8], EV-C117 [9], EV-C118 [10], CV-A21 |
| EV-D | – | EV-D68 |
2. Overview of Rhinovirus Biology
2.1. Brief Overview of Basic Virology
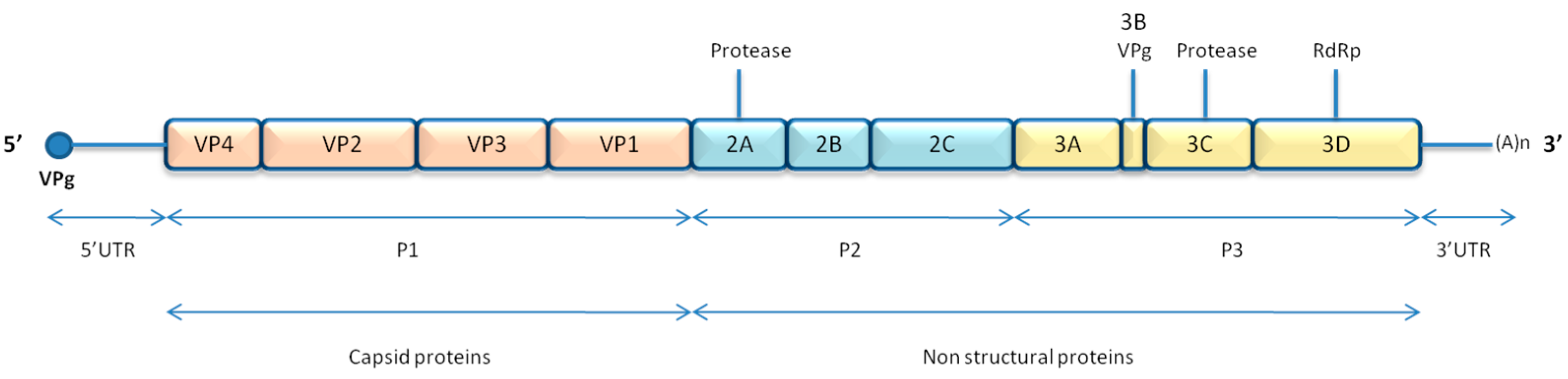
2.1.1. Genome and Structure
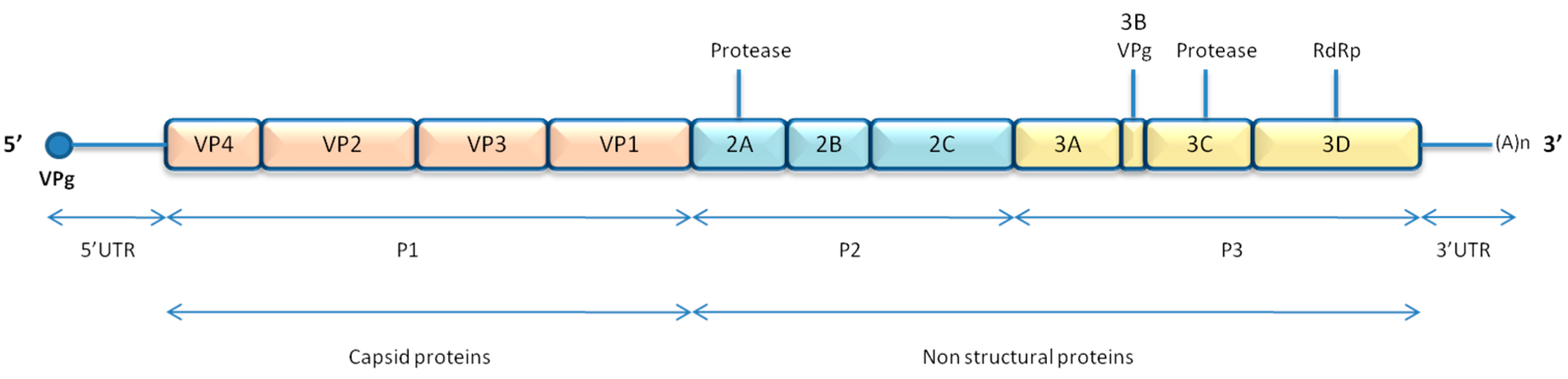
2.1.2. Replication Cycle
2.2. Pathogenesis and Associated Diseases
2.2.1. Transmission
2.2.2. Site of Infection and Pathogenesis
| NON-RV Respiratory Enteroviruses | Rhinoviruses | ||||
|---|---|---|---|---|---|
| Species | Genotype | Receptor | Species | Genotype | Receptor |
| EV-A | CV-A10 | unknown | RV-A | A7, A8, A9, A10, A11, A12, A13, A15, A16, A18, A19, A20, A21, A22, A24, A27, A28, A32, A33, A34, A36, A38, A39, A40, A41, A43, A45, A46, A50, A51, A53, A54, A55, A56, A57, A58, A59, A60, A61, A63, A64, A65, A66, A67, A68, A71, A73, A74, A75, A76, A77, A78, A80, A81, A82, A85, A88, A89, A90, A94, A96, A100, A101, A102, A103, A104, A105, A106, A107, A108, A109 | ICAM-1 [25] |
| CV-A16 | SCARB2 [26], PSGL-1 [27] | ||||
| EV-A71 | SCARB2 [26], PSGL-1 [27] * | ||||
| EV-B | CV-A9 | αV integrins [28,29,30] | |||
| CV-B1 | CAR [31,32], DAF [33] | ||||
| CV-B2 | CAR [31,32] | ||||
| CV-B3 | CAR [31,32], DAF [33] | ||||
| CV-B4 | CAR [31,32] | ||||
| CV-B5 | CAR [31,32], DAF [33] | ||||
| CV-B6 | CAR [31,32] | ||||
| E-1 | α2β1 integrin [34] | ||||
| E-2 | unknown | ||||
| E-3 | DAF [35] | ||||
| E-4 | unknown | ||||
| E-5 | Heparan sulfate [36] | ||||
| E-6 | DAF [35,37] | ||||
| E-7 | DAF [35,37,38] | ||||
| E-9 | αvβ3 integrin [39] | ||||
| E-11 | DAF [35,37], HLA Class I [40] | ||||
| E-12 | DAF [35,37] | ||||
| E-13 | DAF [38] | ||||
| E-14 | unknown | ||||
| E-15 | unknown | A1A, A1B, A2, A23, A25, A29, A30, A31, A44, A47, A49, A62 | LDLR, VLDLR, LRP [41,42,43] | ||
| E-16 | unknown | ||||
| E-17 | unknown | ||||
| E-18 | unknown | ||||
| E-19 | DAF [35] | ||||
| E-20 | DAF [37] | ||||
| E-21 | DAF [37,38] | ||||
| E-25 | DAF [35] | ||||
| E-29 | DAF [38] | ||||
| E-30 | DAF [35] | ||||
| EV-C | CV-A21 | ICAM-1 [44], DAF [45] | RV-B | B3, B4, B5, B6, B14, B17, B26, B27, B35, B37, B42, B48, B52, B69, B70, B72, B79, B83, B84, B86, B91, B92, B93, B97, B99, B100, B101, B102, B103, B104, B105, B106 | ICAM-1 [25] |
| CV-A24 | unknown | ||||
| CV-A24v | Sialic acid [46] | ||||
| EV-C104 | unknown | ||||
| EV-C105 | unknown | ||||
| EV-C109 | unknown | ||||
| EV-C117 | unknown | ||||
| EC-C118 | unknown | ||||
| PV-3 | PVR [47] | ||||
| EV-D | EV-D68 | α2-6-linked sialic acids [48] | RV-C | C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11, C12, C13, C14, C15, C16, C17, C18, C19, C20, C21, C22, C23, C24, C25, C26, C27, C28, C29, C30, C31, C32, C33, C34, C35, C36, C37, C38, C39, C40, C41, C42, C43, C44, C45, C46, C47, C48, C49, C50, C51, C52, C53, C54, C55 | CDHR3 [49] |
2.2.3. Host Response
2.2.4. Clinical Syndromes and Epidemiology
2.2.5. Animal Models
3. Update on Latest Findings on RVs and Respiratory EVs
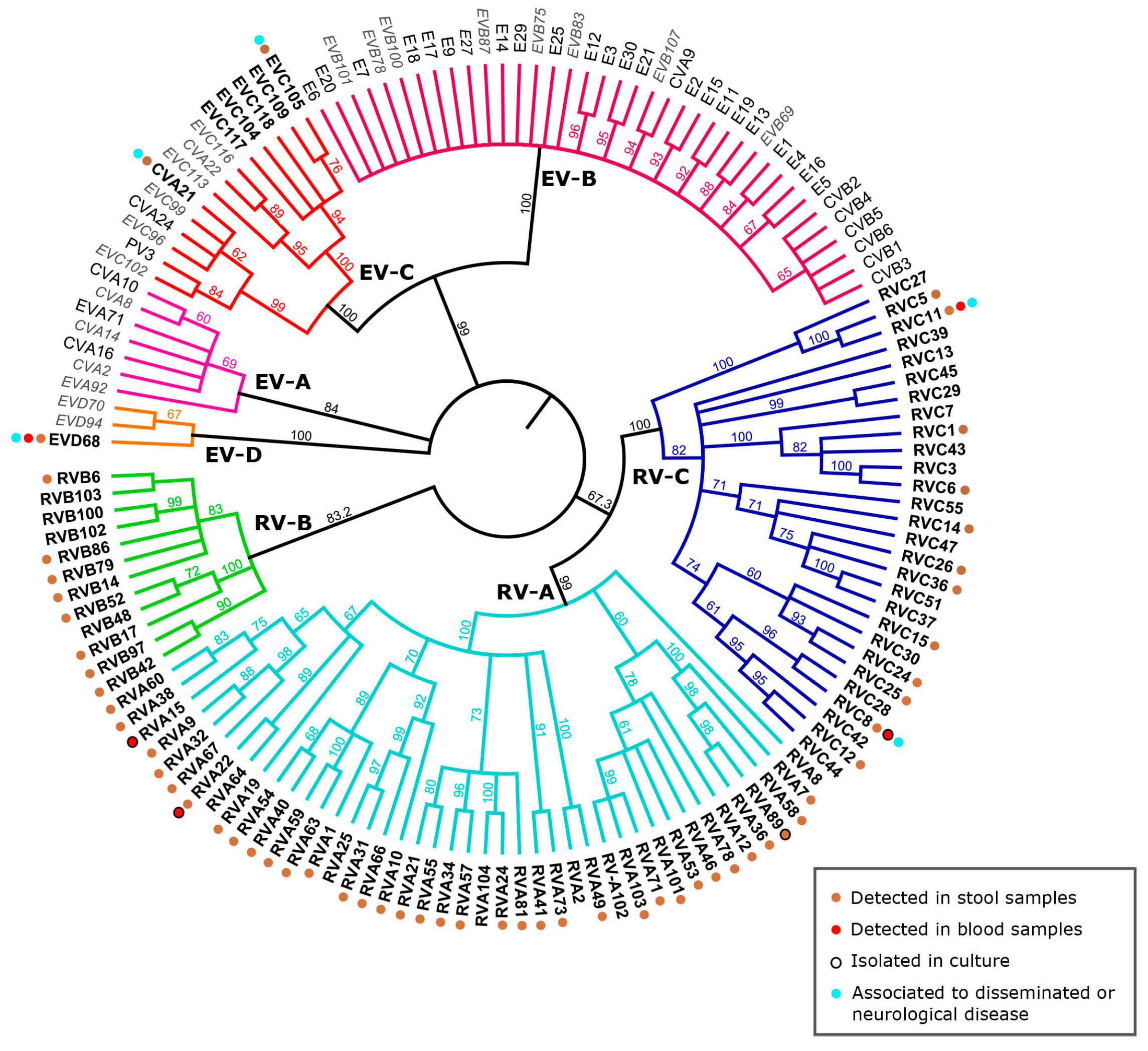
3.1. RV and EV Classification: Current Status

3.2. Mechanisms of RV Evolution and Adaptation
3.3. Recent Advances in Knowledge of the In Vitro and in Vivo Pathogenesis of RV and Respiratory EV
3.3.1. RV-C: Getting to Know the Newcomers
3.3.2. Recent Re-Emergence of EV-D68 and Other Respiratory EVs
3.4. Optimal Growth Temperature of RV: A Possible Link with the Interferon Response
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Makela, M.J.; Puhakka, T.; Ruuskanen, O.; Leinonen, M.; Saikku, P.; Kimpimaki, M.; Blomqvist, S.; Hyypia, T.; Arstila, P. Viruses and bacteria in the etiology of the common cold. J. Clin. Microbiol. 1998, 36, 539–542. [Google Scholar] [PubMed]
- Fendrick, A.M.; Monto, A.S.; Nightengale, B.; Sarnes, M. The economic burden of non-influenza-related viral respiratory tract infection in the united states. Arch. Intern. Med. 2003, 163, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet.Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.R.; Newland, J.G. Viral meningitis and encephalitis: Traditional and emerging viral agents. Semin. Pediatr. Infect. Dis. 2003, 14, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Chapter 17: Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses, and Newer Enteroviruses . In Fields Virology, 6th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Tapparel, C.; Junier, T.; Gerlach, D.; Van-Belle, S.; Turin, L.; Cordey, S.; Muhlemann, K.; Regamey, N.; Aubert, J.D.; Soccal, P.M.; et al. New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerg. Infect. Dis. 2009, 15, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Grard, G.; Drexler, J.F.; Lekana-Douki, S.; Caron, M.; Lukashev, A.; Nkoghe, D.; Gonzalez, J.P.; Drosten, C.; Leroy, E. Type 1 wild poliovirus and putative enterovirus 109 in an outbreak of acute flaccid paralysis in Congo, October-November 2010. Euro Surveil. Eur. Commun. Dis. Bull. 2010, 15, 47. [Google Scholar]
- Yozwiak, N.L.; Skewes-Cox, P.; Gordon, A.; Saborio, S.; Kuan, G.; Balmaseda, A.; Ganem, D.; Harris, E.; DeRisi, J.L. Human enterovirus 109: A novel interspecies recombinant enterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua. J. Virol. 2010, 84, 9047–9058. [Google Scholar] [CrossRef] [PubMed]
- Daleno, C.; Piralla, A.; Scala, A.; Baldanti, F.; Usonis, V.; Principi, N.; Esposito, S. Complete genome sequence of a novel human enterovirus C (HEV-C117) identified in a child with community-acquired pneumonia. J. Virol. 2012, 86, 10888–10889. [Google Scholar] [CrossRef] [PubMed]
- Daleno, C.; Greenberg, D.; Piralla, A.; Scala, A.; Baldanti, F.; Principi, N.; Esposito, S. A novel human enterovirus C (EV-C118) identified in two children hospitalised because of acute otitis media and community-acquired pneumonia in Israel. J. Clin. Virol. 2013, 56, 159–162. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.R.; Walton, R.P.; Shetty, S.; Peel, T.J.; Paktiawal, N.; Kebadze, T.; Gogsadze, L.; Niespodziana, K.; Valenta, R.; Bartlett, N.W.; et al. Rhinovirus infections and immunisation induce cross-serotype reactive antibodies to VP1. Antivir. Res. 2012, 95, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I. The genome-linked protein VPG of vertebrate viruses—A multifaceted protein. Curr. Opin. Virol. 2011, 1, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Langereis, M.A.; Feng, Q.; Nelissen, F.H.; Virgen-Slane, R.; van Noort, G.J.; Maciejewski, S.; Filippov, D.V.; Semler, B.L.; van Delft, F.L.; van Kuppeveld, F.J. Modification of picornavirus genomic RNA using “click” chemistry shows that unlinking of the VPG peptide is dispensable for translation and replication of the incoming viral RNA. Nucleic Acids Res. 2014, 42, 2473–2482. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.; Blaas, D. Uncoating of human rhinoviruses. Rev. Med. Virol. 2010, 20, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A. Modulation of lipid synthesis and trafficking pathways by picornaviruses. Curr. Opin. Virol. 2014, 9, 19–23. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, L.; Wolthers, K.C.; van Kuppeveld, F.J. Replication and inhibitors of enteroviruses and parechoviruses. Viruses 2015, 7, 4529–4562. [Google Scholar] [CrossRef] [PubMed]
- L'Huillier, A.G.; Tapparel, C.; Turin, L.; Boquete-Suter, P.; Thomas, Y.; Kaiser, L. Survival of rhinoviruses on human fingers. Clin. Microbiol. Infect. 2015, 21, 381–385. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dick, E.C.; Jennings, L.C.; Mink, K.A.; Wartgow, C.D.; Inhorn, S.L. Aerosol transmission of rhinovirus colds. J. Infect. Dis. 1987, 156, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Arruda, E.; Boyle, T.R.; Winther, B.; Pevear, D.C.; Gwaltney, J.M., Jr.; Hayden, F.G. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization. J. Infect. Dis. 1995, 171, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Johnston, S.L. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-κB-mediated transcription. J. Biol. Chem. 1999, 274, 9707–9720. [Google Scholar] [CrossRef] [PubMed]
- Winther, B.; Arruda, E.; Witek, T.J.; Marlin, S.D.; Tsianco, M.M.; Innes, D.J.; Hayden, F.G. Expression of ICAM-1 in nasal epithelium and levels of soluble ICAM-1 in nasal lavage fluid during human experimental rhinovirus infection. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Winther, B.; Gwaltney, J.M.; Hendley, J.O. Respiratory virus infection of monolayer cultures of human nasal epithelial cells. Am. Rev. Respir. Dis. 1990, 141, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [PubMed]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Greve, J.M.; Davis, G.; Meyer, A.M.; Forte, C.P.; Yost, S.C.; Marlor, C.W.; Kamarck, M.E.; McClelland, A. The major human rhinovirus receptor is ICAM-1. Cell 1989, 56, 839–847. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamashita, Y.; Li, J.; Hanagata, N.; Minowa, T.; Takemura, T.; Koike, S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 2009, 15, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Shimojima, M.; Tano, Y.; Miyamura, T.; Wakita, T.; Shimizu, H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Med. 2009, 15, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Roivainen, M.; Hyypia, T.; Piirainen, L.; Kalkkinen, N.; Stanway, G.; Hovi, T. RGD-dependent entry of coxsackievirus A9 into host cells and its bypass after cleavage of VP1 protein by intestinal proteases. J. Virol. 1991, 65, 4735–4740. [Google Scholar] [PubMed]
- Williams, C.H.; Kajander, T.; Hyypia, T.; Jackson, T.; Sheppard, D.; Stanway, G. Integrin αvβ6 is an RGD-dependent receptor for coxsackievirus A9. J. Virol. 2004, 78, 6967–6973. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, O.; Susi, P.; Stanway, G.; Hyypia, T. Integrin αvβ6 is a high-affinity receptor for coxsackievirus A9. J. Gen. Virol. 2009, 90, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Martino, T.A.; Petric, M.; Weingartl, H.; Bergelson, J.M.; Opavsky, M.A.; Richardson, C.D.; Modlin, J.F.; Finberg, R.W.; Kain, K.C.; Willis, N.; et al. The coxsackie-adenovirus receptor (CAR) is used by reference strains and clinical isolates representing all six serotypes of coxsackievirus group B and by swine vesicular disease virus. Virology 2000, 271, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Shafren, D.R.; Bates, R.C.; Agrez, M.V.; Herd, R.L.; Burns, G.F.; Barry, R.D. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995, 69, 3873–3877. [Google Scholar] [PubMed]
- Bergelson, J.M.; Shepley, M.P.; Chan, B.M.; Hemler, M.E.; Finberg, R.W. Identification of the integrin VLA-2 as a receptor for echovirus 1. Science 1992, 255, 1718–1720. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.M.; Schmitt, V.; Ward, T.; Goodfellow, I.; Evans, D.J.; Almond, J.W. Characterization of echoviruses that bind decay accelerating factor (CD55): Evidence that some haemagglutinating strains use more than one cellular receptor. J. Gen. Virol. 1998, 79 Pt 7, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Israelsson, S.; Gullberg, M.; Jonsson, N.; Roivainen, M.; Edman, K.; Lindberg, A.M. Studies of echovirus 5 interactions with the cell surface: Heparan sulfate mediates attachment to the host cell. Virus Res. 2010, 151, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Chan, M.; Solomon, K.R.; St John, N.F.; Lin, H.; Finberg, R.W. Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc. Natl. Acad. Sci. USA 1994, 91, 6245–6248. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.; Pipkin, P.A.; Clarkson, N.A.; Stone, D.M.; Minor, P.D.; Almond, J.W. Decay-accelerating factor CD55 is identified as the receptor for echovirus 7 using celics, a rapid immuno-focal cloning method. EMBO J. 1994, 13, 5070–5074. [Google Scholar] [PubMed]
- Paananen, A.; Ylipaasto, P.; Rieder, E.; Hovi, T.; Galama, J.; Roivainen, M. Molecular and biological analysis of echovirus 9 strain isolated from a diabetic child. J. Med. Virol. 2003, 69, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Chevaliez, S.; Balanant, J.; Maillard, P.; Lone, Y.C.; Lemonnier, F.A.; Delpeyroux, F. Role of class I human leukocyte antigen molecules in early steps of echovirus infection of rhabdomyosarcoma cells. Virology 2008, 381, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Hofer, F.; Gruenberger, M.; Kowalski, H.; Machat, H.; Huettinger, M.; Kuechler, E.; Blaas, D. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1839–1842. [Google Scholar] [CrossRef] [PubMed]
- Marlovits, T.C.; Abrahamsberg, C.; Blaas, D. Very-low-density lipoprotein receptor fragment shed from hela cells inhibits human rhinovirus infection. J. Virol. 1998, 72, 10246–10250. [Google Scholar] [PubMed]
- Marlovits, T.C.; Zechmeister, T.; Gruenberger, M.; Ronacher, B.; Schwihla, H.; Blaas, D. Recombinant soluble low density lipoprotein receptor fragment inhibits minor group rhinovirus infection in vitro. FASEB J. 1998, 12, 695–703. [Google Scholar] [PubMed]
- Shafren, D.R.; Dorahy, D.J.; Greive, S.J.; Burns, G.F.; Barry, R.D. Mouse cells expressing human intercellular adhesion molecule-1 are susceptible to infection by coxsackievirus A21. J. Virol. 1997, 71, 785–789. [Google Scholar] [PubMed]
- Shafren, D.R.; Dorahy, D.J.; Ingham, R.A.; Burns, G.F.; Barry, R.D. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J. Virol. 1997, 71, 4736–4743. [Google Scholar] [PubMed]
- Nilsson, E.C.; Jamshidi, F.; Johansson, S.M.; Oberste, M.S.; Arnberg, N. Sialic acid is a cellular receptor for coxsackievirus A24 variant, an emerging virus with pandemic potential. J. Virol. 2008, 82, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef]
- Imamura, T.; Okamoto, M.; Nakakita, S.; Suzuki, A.; Saito, M.; Tamaki, R.; Lupisan, S.; Roy, C.N.; Hiramatsu, H.; Sugawara, K.E.; et al. Antigenic and receptor binding properties of enterovirus 68. J. Virol. 2014, 88, 2374–2384. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Watters, K.; Ashraf, S.; Griggs, T.F.; Devries, M.K.; Jackson, D.J.; Palmenberg, A.C.; Gern, J.E. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus c binding and replication. Proc. Natl. Acad. Sci. USA 2015, 112, 5485–5490. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.X.; Ma, L.; Liu, Q.W.; Li, C.; Huang, Z.; Wu, L.; Xiong, S.D.; Wang, J.H.; Wang, H.B. The molecule of DC-sign captures enterovirus 71 and confers dendritic cell-mediated viral trans-infection. Virol. J. 2014, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chuang, H.; Yang, K.D. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol. J. 2009, 6. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.W.; Poh, C.L.; Sam, I.C.; Chan, Y.F. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J. Virol. 2013, 87, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Su, P.Y.; Wang, Y.F.; Huang, S.W.; Lo, Y.C.; Wang, Y.H.; Wu, S.R.; Shieh, D.B.; Chen, S.H.; Wang, J.R.; Lai, M.D.; et al. Cell surface nucleolin facilitates enterovirus 71 binding and infection. J. Virol. 2015, 89, 4527–4538. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Cong, H.; Tian, H.; Zhang, H.; Zhang, W.; Song, L.; Tien, P. Cell surface vimentin is an attachment receptor for enterovirus 71. J. Virol. 2014, 88, 5816–5833. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Chou, Y.T.; Wu, C.N.; Ho, M.S. Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J. Virol. 2011, 85, 11809–11820. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.A.; Parsons, R. Some virus isolations from common colds: III. Cytopathic effects in tissue cultures. Lancet 1960, 1, 239–242. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Bates, P.J.; Bardin, P.G.; Papi, A.; Leir, S.H.; Fraenkel, D.J.; Meyer, J.; Lackie, P.M.; Sanderson, G.; Holgate, S.T.; et al. Rhinoviruses infect the lower airways. J. Infect. Dis. 2000, 181, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.G.; Sanderson, G.; Hunter, J.; Johnston, S.L. Rhinoviruses replicate effectively at lower airway temperatures. J. Med. Virol. 1999, 58, 100–104. [Google Scholar] [CrossRef]
- Tapparel, C.; Sobo, K.; Constant, S.; Huang, S.; Van Belle, S.; Kaiser, L. Growth and characterization of different human rhinovirus C types in three-dimensional human airway epithelia reconstituted in vitro. Virology 2013, 446, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E.; Galagan, D.M.; Jarjour, N.N.; Dick, E.C.; Busse, W.W. Detection of rhinovirus rna in lower airway cells during experimentally induced infection. Am. J. Respir. Crit. Care Med. 1997, 155, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Pitkaranta, A.; Arruda, E.; Malmberg, H.; Hayden, F.G. Detection of rhinovirus in sinus brushings of patients with acute community-acquired sinusitis by reverse transcription-PCR. J. Clin. Microbiol. 1997, 35, 1791–1793. [Google Scholar] [PubMed]
- Jang, Y.J.; Kwon, H.J.; Park, H.W.; Lee, B.J. Detection of rhinovirus in turbinate epithelial cells of chronic sinusitis. Am. J. Rhinol. 2006, 20, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Chantzi, F.M.; Papadopoulos, N.G.; Bairamis, T.; Tsiakou, M.; Bournousouzis, N.; Constantopoulos, A.G.; Liapi, G.; Xatzipsalti, M.; Kafetzis, D.A. Human rhinoviruses in otitis media with effusion. Pediatr. Allergy Immunol. 2006, 17, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Lupo, J.; Schuffenecker, I.; Morel-Baccard, C.; Bardet, J.; Payen, V.; Kaiser, L.; Constant, S.; Lobrinus, J.A.; Lin-Marq, N.; Lina, B.; et al. Disseminated rhinovirus C8 infection with infectious virus in blood and fatal outcome in a child with repeated episodes of bronchiolitis. J. Clin. Microbiol. 2015, 53, 1775–1777. [Google Scholar] [CrossRef] [PubMed]
- Harvala, H.; McIntyre, C.L.; McLeish, N.J.; Kondracka, J.; Palmer, J.; Molyneaux, P.; Gunson, R.; Bennett, S.; Templeton, K.; Simmonds, P. High detection frequency and viral loads of human rhinovirus species a to C in fecal samples; diagnostic and clinical implications. J. Med. Virol. 2012, 84, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Daleno, C.; Scala, A.; Castellazzi, L.; Terranova, L.; Sferrazza Papa, S.; Longo, M.R.; Pelucchi, C.; Principi, N. Impact of rhinovirus nasopharyngeal viral load and viremia on severity of respiratory infections in children. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, G.E.; Stott, E.J. Rhinoviraemia. Br. Med. J. 1970, 4, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, H.; Oikarinen, S.; Peltonen, P.; Simell, O.; Ilonen, J.; Veijola, R.; Knip, M.; Hyoty, H. Human rhinoviruses including group C are common in stool samples of young Finnish children. J. Clin. Virol. 2013, 56, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Souza, N.; Dolganov, G.; Dubin, R.; Sachs, L.A.; Sassina, L.; Sporer, H.; Yagi, S.; Schnurr, D.; Boushey, H.A.; Widdicombe, J.H. Resistance of differentiated human airway epithelium to infection by rhinovirus. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L373–L381. [Google Scholar] [CrossRef] [PubMed]
- Korpi-Steiner, N.L.; Bates, M.E.; Lee, W.M.; Hall, D.J.; Bertics, P.J. Human rhinovirus induces robust ip-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and stat1 activation. J. Leukocyte Biol. 2006, 80, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Hatchwell, L.; Collison, A.; Girkin, J.; Parsons, K.; Li, J.; Zhang, J.; Phipps, S.; Knight, D.; Bartlett, N.W.; Johnston, S.L.; et al. Toll-like receptor 7 governs interferon and inflammatory responses to rhinovirus and is suppressed by IL-5-induced lung eosinophilia. Thorax 2015, 70, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Vakakis, E.; Richer, E.A.; Evans, G.L.; Villiers, J.P.; Triantafilou, M. Human rhinovirus recognition in non-immune cells is mediated by toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence 2011, 2, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Hewson, C.A.; Jardine, A.; Edwards, M.R.; Laza-Stanca, V.; Johnston, S.L. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 2005, 79, 12273–12279. [Google Scholar] [CrossRef] [PubMed]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberge, P.B.; van Kempen, M.J.; Bachert, C. The common cold at the turn of the millennium. Am. J. Rhinol. 2000, 14, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Barclay, W.S.; al-Nakib, W.; Higgins, P.G.; Tyrrell, D.A. The time course of the humoral immune response to rhinovirus infection. Epidemiol. Infect. 1989, 103, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Alper, C.M.; Doyle, W.J.; Skoner, D.P.; Buchman, C.A.; Seroky, J.T.; Gwaltney, J.M.; Cohen, S.A. Prechallenge antibodies: Moderators of infection rate, signs, and symptoms in adults experimentally challenged with rhinovirus type 39. Laryngoscope 1996, 106, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Glanville, N.; McLean, G.R.; Guy, B.; Lecouturier, V.; Berry, C.; Girerd, Y.; Gregoire, C.; Walton, R.P.; Pearson, R.M.; Kebadze, T.; et al. Cross-serotype immunity induced by immunization with a conserved rhinovirus capsid protein. PLoS Pathog. 2013, 9, e1003669. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Contoli, M. Rhinovirus vaccination: The case against. Eur. Respiratory J. 2011, 37, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.B. Epidemiology, pathogenesis, and treatment of the common cold. Ann. Allergy Asthma Immunol. 1997, 78, 531–539. [Google Scholar] [CrossRef]
- Lessler, J.; Reich, N.G.; Brookmeyer, R.; Perl, T.M.; Nelson, K.E.; Cummings, D.A. Incubation periods of acute respiratory viral infections: A systematic review. Lancet Infect. Dis. 2009, 9, 291–300. [Google Scholar] [CrossRef]
- Pappas, D.E.; Hendley, J.O.; Hayden, F.G.; Winther, B. Symptom profile of common colds in school-aged children. Pediatr. Infect. Dis. J. 2008, 27, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S. The seasonality of rhinovirus infections and its implications for clinical recognition. Clin. Ther. 2002, 24, 1987–1997. [Google Scholar] [CrossRef]
- Winther, B.; Hayden, F.G.; Hendley, J.O. Picornavirus infections in children diagnosed by RT-PCR during longitudinal surveillance with weekly sampling: Association with symptomatic illness and effect of season. J. Med. Virol. 2006, 78, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Linder, J.E.; Kraft, D.C.; Mohamed, Y.; Lu, Z.; Heil, L.; Tollefson, S.; Saville, B.R.; Wright, P.F.; Williams, J.V.; Miller, E.K. Human rhinovirus C: Age, season, and lower respiratory illness over the past 3 decades. J. Allergy Clin. Immunol. 2013, 131, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.C.; Dick, C.R. A subclinical outbreak of human rhinovirus 31 infection in chimpanzees. Am. J. Epidemiol. 1968, 88, 267–272. [Google Scholar] [PubMed]
- Pinto, C.A.; Haff, R.F. Experimental infection of gibbons with rhinovirus. Nature 1969, 224, 1310–1311. [Google Scholar] [CrossRef] [PubMed]
- Gwaltney, J.M. Clinical significance and pathogenesis of viral respiratory infections. Am. J. Med. 2002, 112, 13S–18S. [Google Scholar] [CrossRef]
- Lau, S.K.; Yip, C.C.; Lung, D.C.; Lee, P.; Que, T.L.; Lau, Y.L.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Detection of human rhinovirus C in fecal samples of children with gastroenteritis. J. Clin. Virol. 2012, 53, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; L’Huillier, A.G.; Rougemont, A.L.; Beghetti, M.; Barazzone-Argiroffo, C.; Kaiser, L. Pneumonia and pericarditis in a child with HRV-C infection: A case report. J. Clin. Virol. 2009, 45, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Register, R.B.; Uncapher, C.R.; Naylor, A.M.; Lineberger, D.W.; Colonno, R.J. Human-murine chimeras of ICAM-1 identify amino acid residues critical for rhinovirus and antibody binding. J. Virol. 1991, 65, 6589–6596. [Google Scholar] [PubMed]
- Staunton, D.E.; Gaur, A.; Chan, P.Y.; Springer, T.A. Internalization of a major group human rhinovirus does not require cytoplasmic or transmembrane domains of ICAM-1. J. Immunol. 1992, 148, 3271–3274. [Google Scholar] [PubMed]
- Tuthill, T.J.; Papadopoulos, N.G.; Jourdan, P.; Challinor, L.J.; Sharp, N.A.; Plumpton, C.; Shah, K.; Barnard, S.; Dash, L.; Burnet, J.; et al. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J. Gen. Virol. 2003, 84, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Lomax, N.B.; Yin, F.H. Evidence for the role of the P2 protein of human rhinovirus in its host range change. J. Virol. 1989, 63, 2396–2399. [Google Scholar] [PubMed]
- Bartlett, N.W.; Walton, R.P.; Edwards, M.R.; Aniscenko, J.; Caramori, G.; Zhu, J.; Glanville, N.; Choy, K.J.; Jourdan, P.; Burnet, J.; et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat. Med. 2008, 14, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.C.; Badmanathan, M.; Devi, S.; Leong, K.L.; Cardosa, M.J.; Wong, K.T. Pathologic characterization of a murine model of human enterovirus 71 encephalomyelitis. J. Neuropathol. Exp. Neurol. 2008, 67, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.L.; Racaniello, V.R. Selection of rhinovirus 1A variants adapted for growth in mouse lung epithelial cells. Virology 2011, 420, 82–88. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Picornaviridae website. Available online: Http://www.Picornaviridae.Com/ (accessed on 08 August 2015).
- Blomqvist, S.; Savolainen, C.; Raman, L.; Roivainen, M.; Hovi, T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J. Clin.Microbiol. 2002, 40, 4218–4223. [Google Scholar] [CrossRef] [PubMed]
- Hamparian, V.V.; Colonno, R.J.; Cooney, M.K.; Dick, E.C.; Gwaltney, J.M., Jr.; Hughes, J.H.; Jordan, W.S., Jr.; Kapikian, A.Z.; Mogabgab, W.J.; Monto, A.; et al. A collaborative report: Rhinoviruses—Extension of the numbering system from 89 to 100. Virology 1987, 159, 191–192. [Google Scholar] [PubMed]
- Rhinoviruses: A numbering system. Nature 1967, 213, 761–762.
- Oberste, M.S.; Nix, W.A.; Maher, K.; Pallansch, M.A. Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J. Clin. Virol. 2003, 26, 375–377. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [PubMed]
- Simmonds, P.; McIntyre, C.; Savolainen-Kopra, C.; Tapparel, C.; Mackay, I.M.; Hovi, T. Proposals for the classification of human rhinovirus species C into genotypically assigned types. J. Gen. Virol. 2010, 91, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Cordey, S.; Junier, T.; Gerlach, D.; Gobbini, F.; Farinelli, L.; Zdobnov, E.M.; Winther, B.; Tapparel, C.; Kaiser, L. Rhinovirus genome evolution during experimental human infection. PLoS ONE 2010, 5, e10588. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Cordey, S.; Junier, T.; Farinelli, L.; Van Belle, S.; Soccal, P.M.; Aubert, J.D.; Zdobnov, E.; Kaiser, L. Rhinovirus genome variation during chronic upper and lower respiratory tract infections. PLoS ONE 2011, 6, e21163. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Bukh, J. Comparative analysis of the molecular mechanisms of recombination in hepatitis C virus. Trends Microbiol. 2014, 22, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Holmblat, B.; Jegouic, S.; Muslin, C.; Blondel, B.; Joffret, M.L.; Delpeyroux, F. Nonhomologous recombination between defective poliovirus and coxsackievirus genomes suggests a new model of genetic plasticity for picornaviruses. mBio 2014, 5, e01119–e01114. [Google Scholar] [CrossRef] [PubMed]
- Gallei, A.; Pankraz, A.; Thiel, H.J.; Becher, P. RNA recombination in vivo in the absence of viral replication. J. Virol. 2004, 78, 6271–6281. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Galli, A.; Li, Y.P.; Mikkelsen, L.S.; Gottwein, J.M.; Bukh, J. Productive homologous and non-homologous recombination of hepatitis C virus in cell culture. PLoS Pathog. 2013, 9, e1003228. [Google Scholar] [CrossRef] [PubMed]
- Gmyl, A.P.; Belousov, E.V.; Maslova, S.V.; Khitrina, E.V.; Chetverin, A.B.; Agol, V.I. Nonreplicative RNA recombination in poliovirus. J. Virol. 1999, 73, 8958–8965. [Google Scholar] [PubMed]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length “imprecise” intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef] [PubMed]
- Schibler, M.; Piuz, I.; Hao, W.; Tapparel, C. Chimeric rhinoviruses obtained via genetic engineering or artificially induced recombination are viable only if the polyprotein coding sequence derives from the same species. J. Virol. 2015, 89, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Lashkevich, V.A.; Ivanova, O.E.; Koroleva, G.A.; Hinkkanen, A.E.; Ilonen, J. Recombination in circulating human enterovirus B: Independent evolution of structural and non-structural genome regions. J. Gen. Virol. 2005, 86, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Santti, J.; Hyypia, T.; Kinnunen, L.; Salminen, M. Evidence of recombination among enteroviruses. J. Virol. 1999, 73, 8741–8749. [Google Scholar] [PubMed]
- Simmonds, P.; Welch, J. Frequency and dynamics of recombination within different species of human enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Junier, T.; Gerlach, D.; Cordey, S.; Van Belle, S.; Perrin, L.; Zdobnov, E.M.; Kaiser, L. New complete genome sequences of human rhinoviruses shed light on their phylogeny and genomic features. BMC Genomics 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.L.; McWilliam Leitch, E.C.; Savolainen-Kopra, C.; Hovi, T.; Simmonds, P. Analysis of genetic diversity and sites of recombination in human rhinovirus species C. J. Virol. 2010, 84, 10297–10310. [Google Scholar] [CrossRef] [PubMed]
- Schibler, M.; Gerlach, D.; Martinez, Y.; Belle, S.V.; Turin, L.; Kaiser, L.; Tapparel, C. Experimental human rhinovirus and enterovirus interspecies recombination. J. Gen. Virol. 2012, 93, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Yip, C.C.; Tsoi, H.W.; Lee, R.A.; So, L.Y.; Lau, Y.L.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J. Clin. Microbiol. 2007, 45, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Arden, K.E.; McErlean, P.; Nissen, M.D.; Sloots, T.P.; Mackay, I.M. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J. Med. Virol. 2006, 78, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Kiang, D.; Yagi, S.; Kantardjieff, K.A.; Kim, E.J.; Louie, J.K.; Schnurr, D.P. Molecular characterization of a variant rhinovirus from an outbreak associated with uncommonly high mortality. J. Clin. Virol. 2007, 38, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Gonzalez, R.; Xie, Z.; Xiao, Y.; Liu, J.; Chen, L.; Liu, C.; Zhang, J.; Ren, L.; Vernet, G.; et al. Human rhinovirus C infections mirror those of human rhinovirus a in children with community-acquired pneumonia. J. Clin. Virol. 2010, 49, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lamson, D.; Renwick, N.; Kapoor, V.; Liu, Z.; Palacios, G.; Ju, J.; Dean, A.; St George, K.; Briese, T.; Lipkin, W.I. Masstag polymerase-chain-reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza-like illness in New York state during 2004–2005. J. Infect. Dis. 2006, 194, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Kiesner, C.; Pappas, T.; Lee, I.; Grindle, K.; Jartti, T.; Jakiela, B.; Lemanske, R.F., Jr.; Shult, P.A.; Gern, J.E. A diverse group of previously unrecognized human rhinoviruses are common causes of respiratory illnesses in infants. PLoS ONE 2007, 2, e966. [Google Scholar] [CrossRef] [PubMed]
- McErlean, P.; Shackelton, L.A.; Lambert, S.B.; Nissen, M.D.; Sloots, T.P.; Mackay, I.M. Characterisation of a newly identified human rhinovirus, HRV-QPM, discovered in infants with bronchiolitis. J. Clin. Virol. 2007, 39, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kistler, A.; Avila, P.C.; Rouskin, S.; Wang, D.; Ward, T.; Yagi, S.; Schnurr, D.; Ganem, D.; DeRisi, J.L.; Boushey, H.A. Pan-viral screening of respiratory tract infections in adults with and without asthma reveals unexpected human coronavirus and human rhinovirus diversity. J. Infect. Dis. 2007, 196, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Renwick, N.; Venter, M.; Jarman, R.G.; Ghosh, D.; Kondgen, S.; Shrestha, S.K.; Hoegh, A.M.; Casas, I.; Adjogoua, E.V.; et al. Global distribution of novel rhinovirus genotype. Emerg. Infect. Dis. 2008, 14, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Lemanske, R.F., Jr.; Evans, M.D.; Vang, F.; Pappas, T.; Gangnon, R.; Jackson, D.J.; Gern, J.E. Human rhinovirus species and season of infection determine illness severity. Am. J. Respir. Crit. Care Med. 2012, 186, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.B.; Wo, Y.; Wang, L.Y.; Wang, H.Y.; Huang, D.D.; Zhang, X.A.; Liu, W.; Cao, W.C. Molecular epidemiology of human rhinovirus in children with acute respiratory diseases in Chongqing, China. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Xatzipsalti, M.; Kyrana, S.; Tsolia, M.; Psarras, S.; Bossios, A.; Laza-Stanca, V.; Johnston, S.L.; Papadopoulos, N.G. Rhinovirus viremia in children with respiratory infections. Am. J. Respir. Crit. Care Med. 2005, 172, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Khetsuriani, N.; Lu, X.; Teague, W.G.; Kazerouni, N.; Anderson, L.J.; Erdman, D.D. Novel human rhinoviruses and exacerbation of asthma in children. Emerg. Infect. Dis. 2008, 14, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.; Kutkowska, A.E.; McWilliam Leitch, E.C.; Gaunt, E.; Templeton, K.; Harvala, H.; Simmonds, P. Genetics, recombination and clinical features of human rhinovirus species C (HRV-C) infections; interactions of HRV-C with other respiratory viruses. PLoS ONE 2009, 4, e8518. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.K.; Khuri-Bulos, N.; Williams, J.V.; Shehabi, A.A.; Faouri, S.; Al Jundi, I.; Chen, Q.; Heil, L.; Mohamed, Y.; Morin, L.L.; et al. Human rhinovirus C associated with wheezing in hospitalised children in the Middle East. J. Clin. Virol. 2009, 46, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Fuji, N.; Suzuki, A.; Lupisan, S.; Sombrero, L.; Galang, H.; Kamigaki, T.; Tamaki, R.; Saito, M.; Aniceto, R.; Olveda, R.; et al. Detection of human rhinovirus C viral genome in blood among children with severe respiratory infections in the Philippines. PLoS ONE 2011, 6, e27247. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yuan, X.H.; Xie, Z.P.; Gao, H.C.; Song, J.R.; Zhang, R.F.; Xu, Z.Q.; Zheng, L.S.; Hou, Y.D.; Duan, Z.J. Prevalence and clinical characterization of a newly identified human rhinovirus C species in children with acute respiratory tract infections. J. Clin. Microbiol. 2009, 47, 2895–2900. [Google Scholar] [CrossRef] [PubMed]
- Iwane, M.K.; Prill, M.M.; Lu, X.; Miller, E.K.; Edwards, K.M.; Hall, C.B.; Griffin, M.R.; Staat, M.A.; Anderson, L.J.; Williams, J.V.; et al. Human rhinovirus species associated with hospitalizations for acute respiratory illness in young US children. J. Infect. Dis. 2011, 204, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Espejo, V.; Nelson, M.; Sovero, M.; Villaran, M.V.; Gomez, J.; Barrantes, M.; Sanchez, F.; Comach, G.; Arango, A.E.; et al. Human rhinoviruses and enteroviruses in influenza-like illness in Latin America. Virol. J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Aponte, F.E.; Taboada, B.; Espinoza, M.A.; Arias-Ortiz, M.A.; Monge-Martinez, J.; Rodriguez-Vazquez, R.; Diaz-Hernandez, F.; Zarate-Vidal, F.; Wong-Chew, R.M.; Firo-Reyes, V.; et al. Rhinovirus is an important pathogen in upper and lower respiratory tract infections in Mexican children. Virol. J. 2015, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Arnold, J.C.; Fairchok, M.P.; Danaher, P.J.; McDonough, E.A.; Blair, P.J.; Garcia, J.; Halsey, E.S.; Schofield, C.; Ottolini, M.; et al. Epidemiologic, clinical, and virologic characteristics of human rhinovirus infection among otherwise healthy children and adults: Rhinovirus among adults and children. J. Clin. Virol. 2015, 64, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Piralla, A.; Rovida, F.; Campanini, G.; Rognoni, V.; Marchi, A.; Locatelli, F.; Gerna, G. Clinical severity and molecular typing of human rhinovirus C strains during a fall outbreak affecting hospitalized patients. J. Clin. Virol. 2009, 45, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Mack, I.; Tapparel, C.; Kaiser, L.; Alves, M.P.; Kieninger, E.; Frey, U.; Regamey, N.; Latzin, P. Human rhinovirus types and association with respiratory symptoms during the first year of life. Pediatr. Infect. Dis. J. 2015, 34, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Zampiero, A.; Gambino, M.; Scala, A.; Senatore, L.; Lelii, M.; Ascolese, B.; Pelucchi, C.; Esposito, S. Prospective evaluation of rhinovirus infection in healthy young children. J. Clin. Virol. 2015, 66, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Palmenberg, A.C.; Lee, W.M.; Rathe, J.A.; Amineva, S.P.; Sun, X.; Pasic, T.R.; Jarjour, N.N.; Liggett, S.B.; Gern, J.E. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat. Med. 2011, 17, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Bernard, K.; Patel, N.; Ulbrandt, N.; Feng, H.; Svabek, C.; Wilson, S.; Stracener, C.; Wang, K.; Suzich, J.; et al. Infection and propagation of human rhinovirus c in human airway epithelial cells. J. Virol. 2012, 86, 13524–13532. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, K.; Bochkov, Y.A.; Ashraf, S.; Brockman-Schneider, R.A.; Evans, M.D.; Pasic, T.R.; Gern, J.E. Effects of rhinovirus species on viral replication and cytokine production. J. Allergy Clin. Immunol. 2014, 134, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Bonnelykke, K.; Sleiman, P.; Nielsen, K.; Kreiner-Moller, E.; Mercader, J.M.; Belgrave, D.; den Dekker, H.T.; Husby, A.; Sevelsted, A.; Faura-Tellez, G.; et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 2014, 46, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Schieble, J.H.; Fox, V.L.; Lennette, E.H. A probable new human picornavirus associated with respiratory diseases. Am. J. Epidemiol. 1967, 85, 297–310. [Google Scholar] [PubMed]
- Oberste, M.S.; Maher, K.; Schnurr, D.; Flemister, M.R.; Lovchik, J.C.; Peters, H.; Sessions, W.; Kirk, C.; Chatterjee, N.; Fuller, S.; et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J. Gen. Virol. 2004, 85, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Hirano, R.; Okamoto-Nakagawa, R.; Ichiyama, T.; Shirabe, K. Enterovirus 68 infection in children with asthma attacks: Virus-induced asthma in japanese children. Allergy 2011, 66, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Fuji, N.; Suzuki, A.; Tamaki, R.; Saito, M.; Aniceto, R.; Galang, H.; Sombrero, L.; Lupisan, S.; Oshitani, H. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg. Infect. Dis. 2011, 17, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.; van der Sanden, S.; Snijders, B.E.; Jaramillo-Gutierrez, G.; Bont, L.; van der Ent, C.K.; Overduin, P.; Jenny, S.L.; Jusic, E.; van der Avoort, H.G.; et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in the Netherlands in 2010. Virology 2012, 423, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Kapoor, V.; Wu, W.; Lurio, J.; Jain, K.; Mostashari, F.; Briese, T.; Lipkin, W.I. Longitudinal molecular microbial analysis of influenza-like illness in New York city, May 2009 through May 2010. Virol. J. 2011, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- CDC Report: Enterovirus D68 in the United States. 2014. Available online: http://www.Cdc.Gov/non-polio-enterovirus/about/ev-d68.Html (Accessed on 8 August 2015).
- Ikeda, T.; Mizuta, K.; Abiko, C.; Aoki, Y.; Itagaki, T.; Katsushima, F.; Katsushima, Y.; Matsuzaki, Y.; Fuji, N.; Imamura, T.; et al. Acute respiratory infections due to enterovirus 68 in yamagata, Japan between 2005 and 2010. Microbiol. Immunol. 2012, 56, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J.D.; Barnes, A.; McCarthy, J.E.; Schwartzman, J.D.; Oberste, M.S.; Rhodes, C.H.; Modlin, J.F.; Wright, P.F. A fatal central nervous system enterovirus 68 infection. Arch. Pathol. Lab. Med. 2011, 135, 793–796. [Google Scholar] [PubMed]
- Khetsuriani, N.; Lamonte-Fowlkes, A.; Oberst, S.; Pallansch, M.A.; Centers for Disease, C. Prevention, Enterovirus surveillance—United States, 1970–2005. Morb. Mortal. Wkly. Rep. 2006, 55, 1–20. [Google Scholar]
- Khan, F. Enterovirus d68: Acute respiratory illness and the 2014 outbreak. Emerg. Med. Clin. N. Am. 2015, 33, e19–e32. [Google Scholar] [CrossRef]
- Pfeiffer, H.C.; Bragstad, K.; Skram, M.K.; Dahl, H.; Knudsen, P.K.; Chawla, M.S.; Holberg-Petersen, M.; Vainio, K.; Dudman, S.G.; Kran, A.M.; et al. Two cases of acute severe flaccid myelitis associated with enterovirus d68 infection in children, Norway, Autumn 2014. Euro Surveil. Eur. Commun. Dis. Bull. 2015, 20, 21062. [Google Scholar] [CrossRef]
- Lang, M.; Mirand, A.; Savy, N.; Henquell, C.; Maridet, S.; Perignon, R.; Labbe, A.; Peigue-Lafeuille, H. Acute flaccid paralysis following enterovirus d68 associated pneumonia, France, 2014. Euro Surveil. Eur. Commun. Dis. Bull. 2014, 19, 44. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Messacar, K.; Clayton, A.; Yu, G.; Somasekar, S.; Federman, S.; Stryke, D.; Anderson, C.; Yagi, S.; et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-2014): A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 671–682. [Google Scholar] [CrossRef]
- Walther, T.; Karamanska, R.; Chan, R.W.; Chan, M.C.; Jia, N.; Air, G.; Hopton, C.; Wong, M.P.; Dell, A.; Malik Peiris, J.S.; et al. Glycomic analysis of human respiratory tract tissues and correlation with influenza virus infection. PLoS Pathog. 2013, 9, e1003223. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.A.; Dimock, K. Sialic acid functions in enterovirus 70 binding and infection. J. Virol. 2002, 76, 11265–11272. [Google Scholar] [CrossRef] [PubMed]
- Mistry, N.; Inoue, H.; Jamshidi, F.; Storm, R.J.; Oberste, M.S.; Arnberg, N. Coxsackievirus A24 variant uses sialic acid-containing O-linked glycoconjugates as cellular receptors on human ocular cells. J. Virol. 2011, 85, 11283–11290. [Google Scholar] [CrossRef] [PubMed]
- Zocher, G.; Mistry, N.; Frank, M.; Hahnlein-Schick, I.; Ekstrom, J.O.; Arnberg, N.; Stehle, T. A sialic acid binding site in a human picornavirus. PLoS Pathog. 2014, 10, e1004401. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Drexler, J.F.; Kotova, V.O.; Amjaga, E.N.; Reznik, V.I.; Gmyl, A.P.; Grard, G.; Taty Taty, R.; Trotsenko, O.E.; Leroy, E.M.; et al. Novel serotypes 105 and 116 are members of distinct subgroups of human enterovirus C. J. Gen. Virol. 2012, 93, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Piralla, A.; Rovida, F.; Baldanti, F.; Gerna, G. Enterovirus genotype EV-104 in humans, Italy, 2008–2009. Emerg. Infect. Dis. 2010, 16, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Kaida, A.; Kubo, H.; Sekiguchi, J.; Hase, A.; Iritani, N. Enterovirus 104 infection in adult, Japan, 2011. Emerg. Infect. Dis. 2012, 18, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Pankovics, P.; Boros, A.; Szabo, H.; Szekely, G.; Gyurkovits, K.; Reuter, G. Human enterovirus 109 (EV109) in acute paediatric respiratory disease in Hungary. Acta Microbiol. Immunol. Hung. 2012, 59, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Hirschberg, D.L.; Sameroff, S.; Haq, S.; Luna, G.; Bennett, A.J.; Silva, M.; Leguia, M.; Kasper, M.; Bausch, D.G.; et al. Genomic analysis of two novel human enterovirus C genotypes found in respiratory samples from Peru. J. Gen. Virol. 2013, 94, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.B.; Wo, Y.; Wang, H.Y.; Zhang, X.A.; Huang, D.D.; Zhao, J.; Liu, E.M.; Liu, W.; Cao, W.C. Detection and complete genome characterization of human enterovirus 118 from children with acute respiratory disease in China. Virus Genes 2014, 48, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Gonzalez, R.; Wang, Z.; Ren, L.; Xiao, Y.; Li, J.; Li, Y.; Vernet, G.; Paranhos-Baccala, G.; Jin, Q.; et al. Coxsackievirus A21, enterovirus 68, and acute respiratory tract infection, China. Emerg. Infect. Dis. 2012, 18, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Bator-Kelly, C.M.; Rieder, E.; Chipman, P.R.; Craig, A.; Kuhn, R.J.; Wimmer, E.; Rossmann, M.G. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure 2005, 13, 1019–1033. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dufresne, A.T.; Gromeier, M. A nonpolio enterovirus with respiratory tropism causes poliomyelitis in intercellular adhesion molecule 1 transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 13636–13641. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, M.; Lin, X.; Liu, Y.; Xiong, P.; Wang, L.; Xu, A.; Tao, Z.; Zhang, D. Molecular characterization of coxsackievirus A21 in shandong, China. Arch. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Chen, E.R.; Hsu, K.H.; Twu, S.J.; Chen, K.T.; Tsai, S.F.; Wang, J.R.; Shih, S.R. An epidemic of enterovirus 71 infection in Taiwan. Taiwan enterovirus epidemic working group. N. Engl. J. Med. 1999, 341, 929–935. [Google Scholar] [CrossRef] [PubMed]
- McMinn, P.; Lindsay, K.; Perera, D.; Chan, H.M.; Chan, K.P.; Cardosa, M.J. Phylogenetic analysis of enterovirus 71 strains isolated during linked epidemics in Malaysia, Singapore, and Western Australia. J. Virol. 2001, 75, 7732–7738. [Google Scholar] [CrossRef] [PubMed]
- Merovitz, L.; Demers, A.M.; Newby, D.; McDonald, J. Enterovirus 71 infections at a Canadian center. Pediatr. Infect. Dis. J. 2000, 19, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.P.; Kuo, P.H.; Liu, C.C.; Wang, J.R. Respiratory viral infections among pediatric inpatients and outpatients in Taiwan from 1997 to 1999. J. Clin. Microbiol. 2001, 39, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.L.; Dickson, K.E.; Waters, M.J.; Kennett, M.L.; Land, S.A.; Sneddon, M. Outbreak of enterovirus 71 infection in Victoria, Australia, with a high incidence of neurologic involvement. Pediatr. Infect. Dis. J. 1988, 7, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.J.; Yang, F.L.; Hsu, Y.H.; Chen, H.I. Mechanism of fulminant pulmonary edema caused by enterovirus 71. Clin. Infect. Dis. 2004, 38, 1784–1788. [Google Scholar] [CrossRef] [PubMed]
- Stott, E.J.; Heath, G.F. Factors affecting the growth of rhinovirus 2 in suspension cultures of L132 cells. J. Gen. Virol. 1970, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.H.; Knight, E., Jr. In vivo and in vitro synthesis of human rhinovirus type 2 ribonucleic acid. J. Virol. 1972, 10, 93–98. [Google Scholar] [PubMed]
- Foxman, E.F.; Storer, J.A.; Fitzgerald, M.E.; Wasik, B.R.; Hou, L.; Zhao, H.; Turner, P.E.; Pyle, A.M.; Iwasaki, A. Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells. Proc. Natl. Acad.Sci. USA 2015, 112, 827–832. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Royston, L.; Tapparel, C. Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC. Viruses 2016, 8, 16. https://doi.org/10.3390/v8010016
Royston L, Tapparel C. Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC. Viruses. 2016; 8(1):16. https://doi.org/10.3390/v8010016
Chicago/Turabian StyleRoyston, Léna, and Caroline Tapparel. 2016. "Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC" Viruses 8, no. 1: 16. https://doi.org/10.3390/v8010016
APA StyleRoyston, L., & Tapparel, C. (2016). Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC. Viruses, 8(1), 16. https://doi.org/10.3390/v8010016