
Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
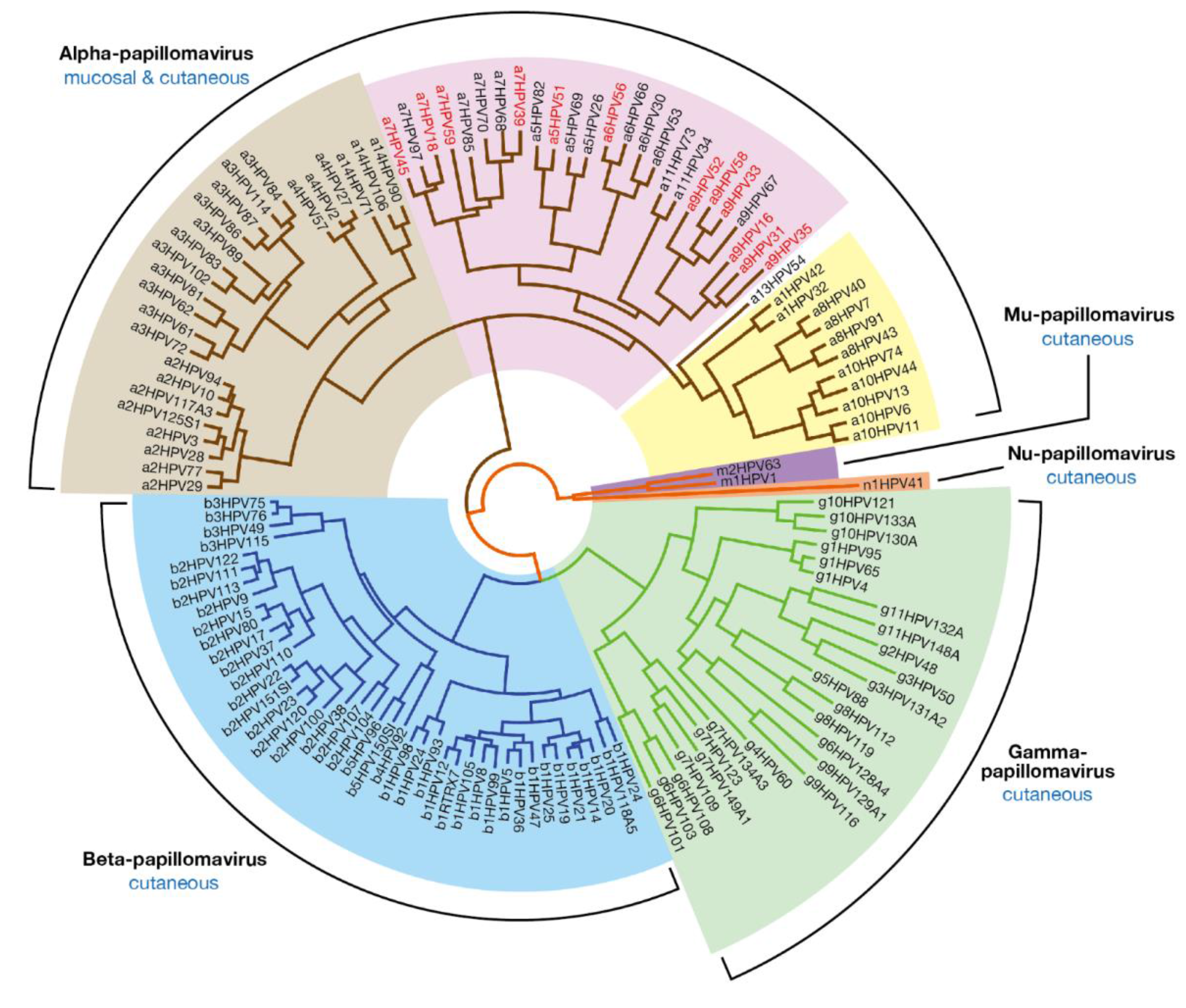
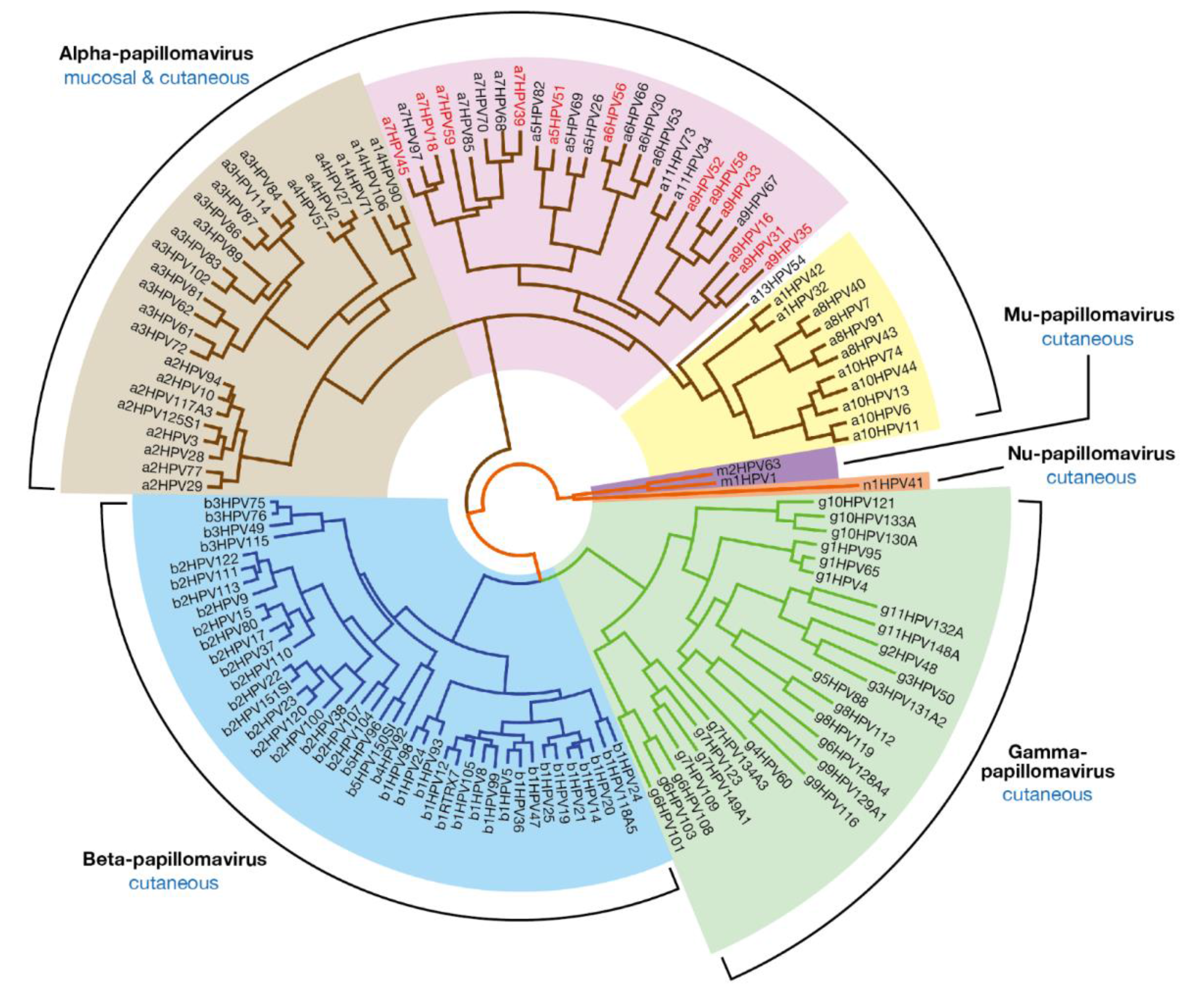
2. Papillomavirus Diversity at the Level of Genotype, Epithelial Tropism and Pathogenicity
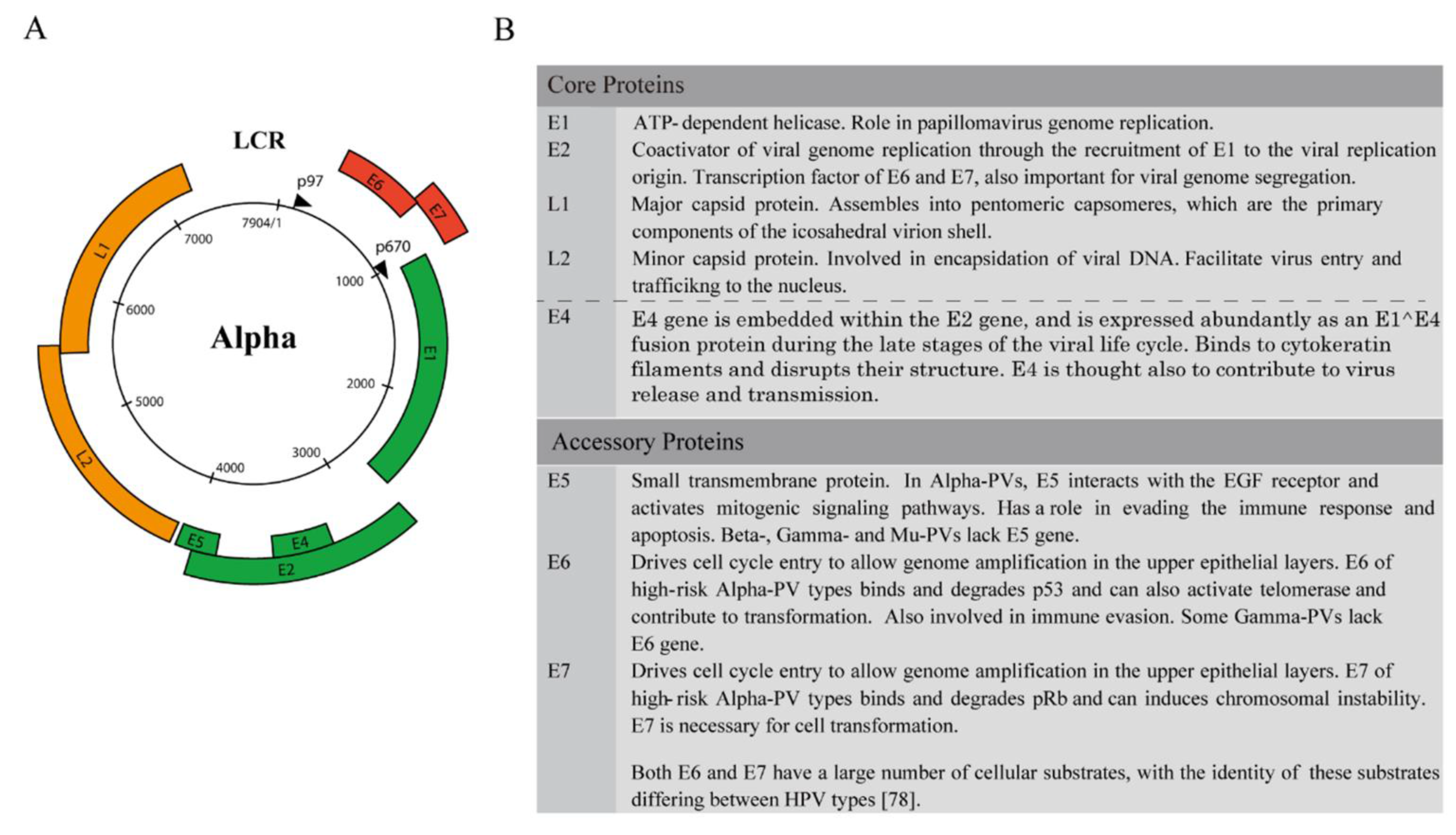
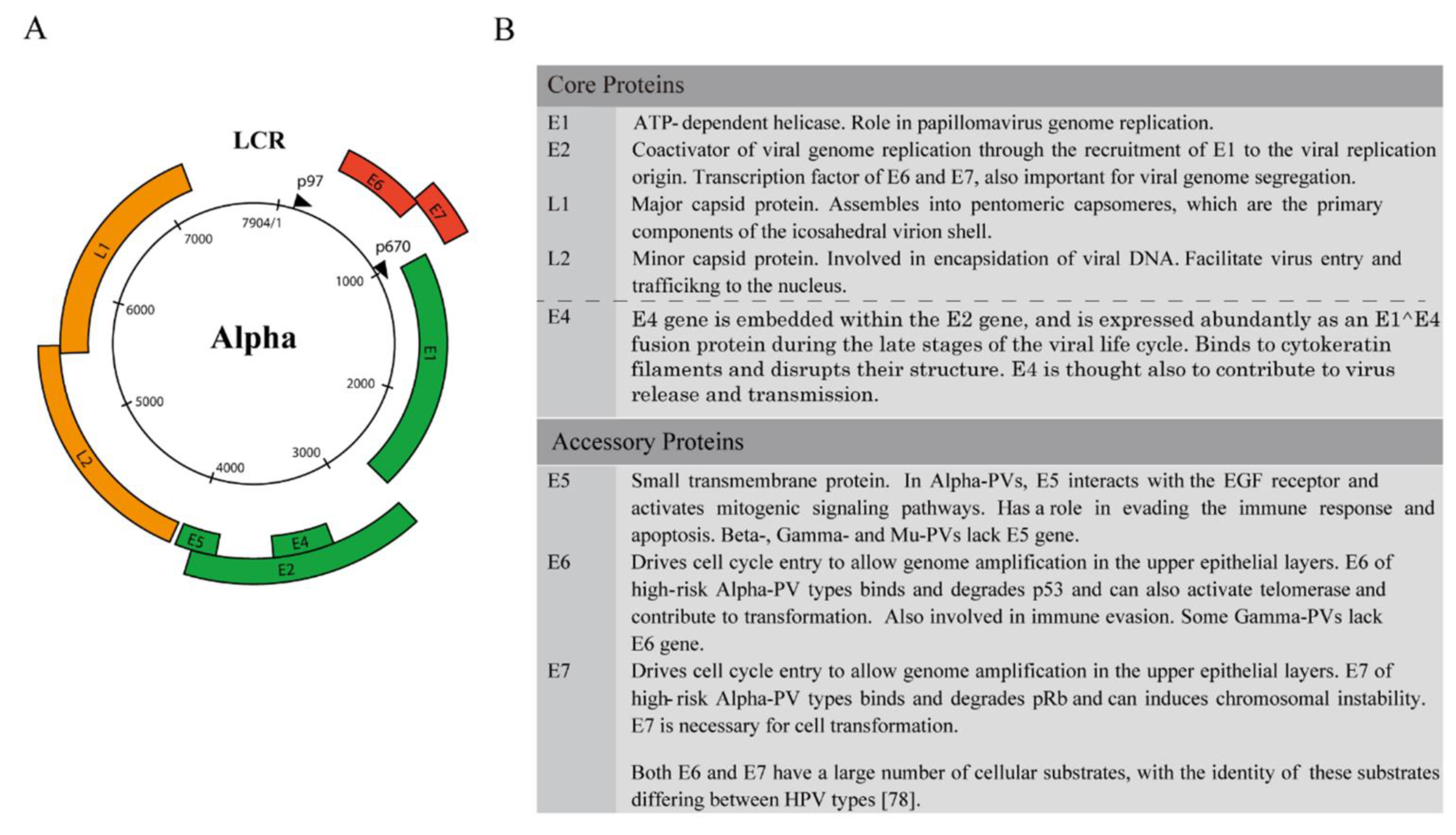
3. Genome Structure and the Classification of Viral Gene Products
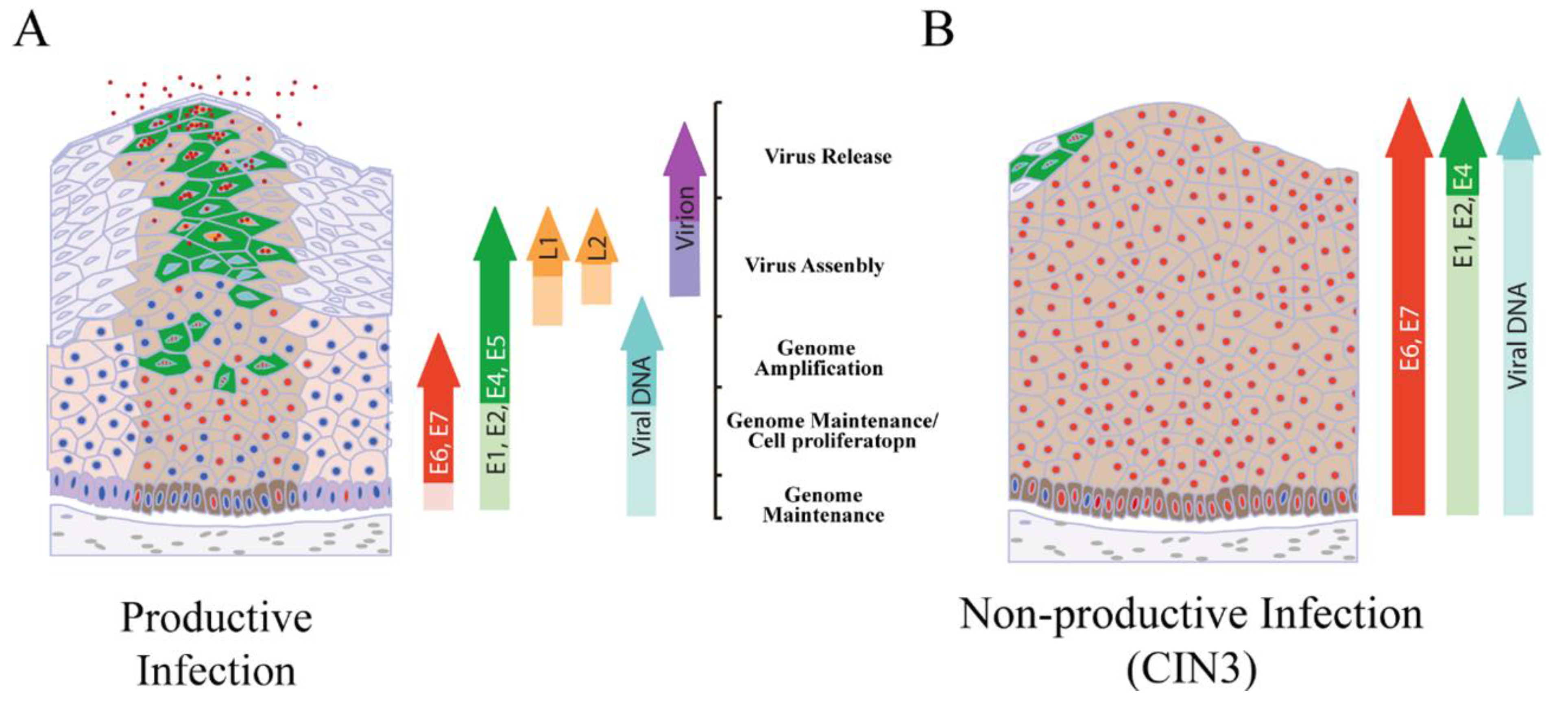
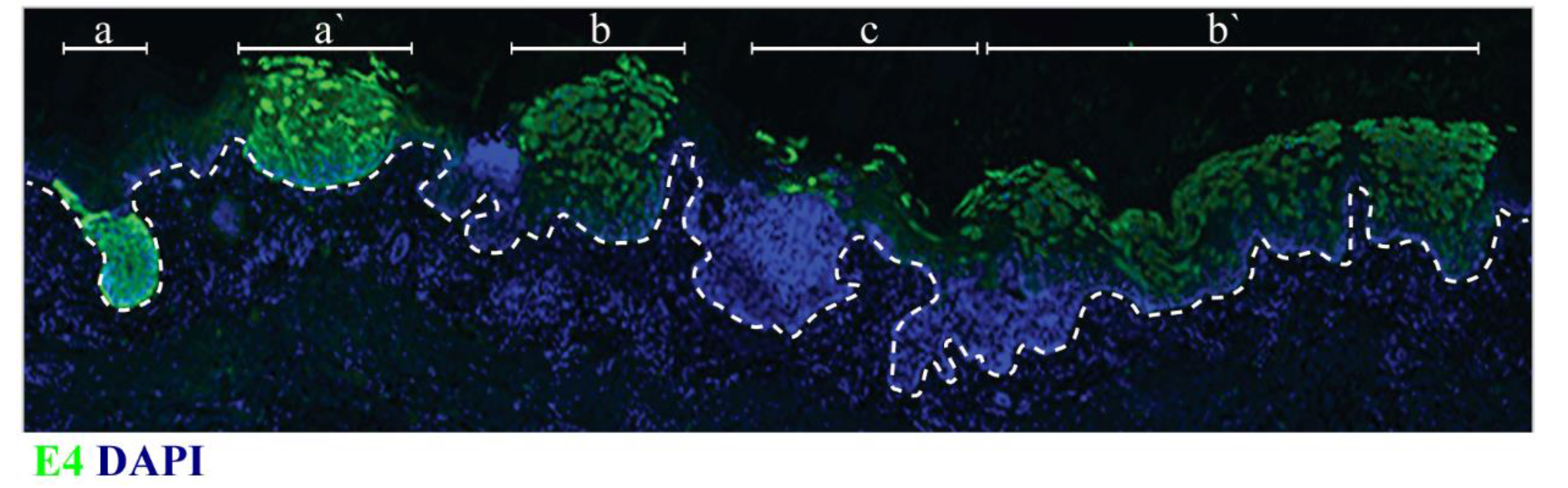
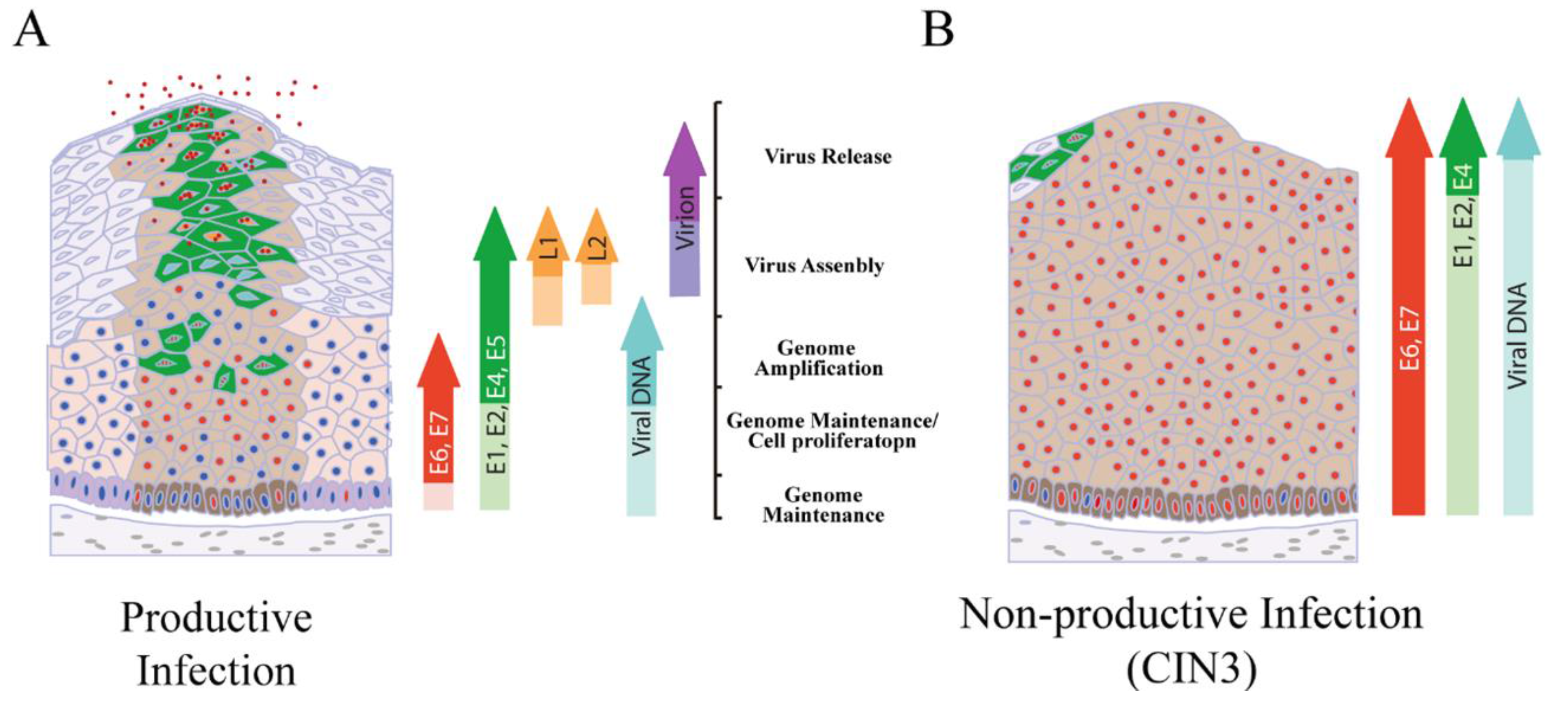
4. Productive Papillomavirus Life Cycle and Non-Productive Infection
5. Epithelial Stem Cells and Their Postulated Role in Papillomavirus-Associated Disease
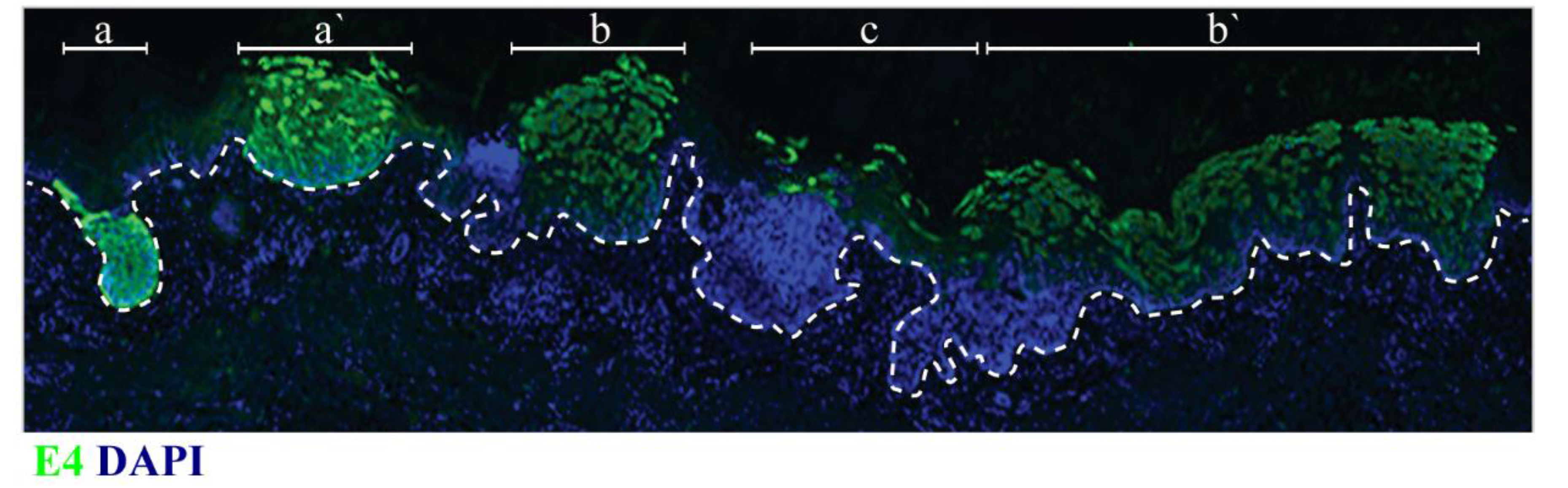
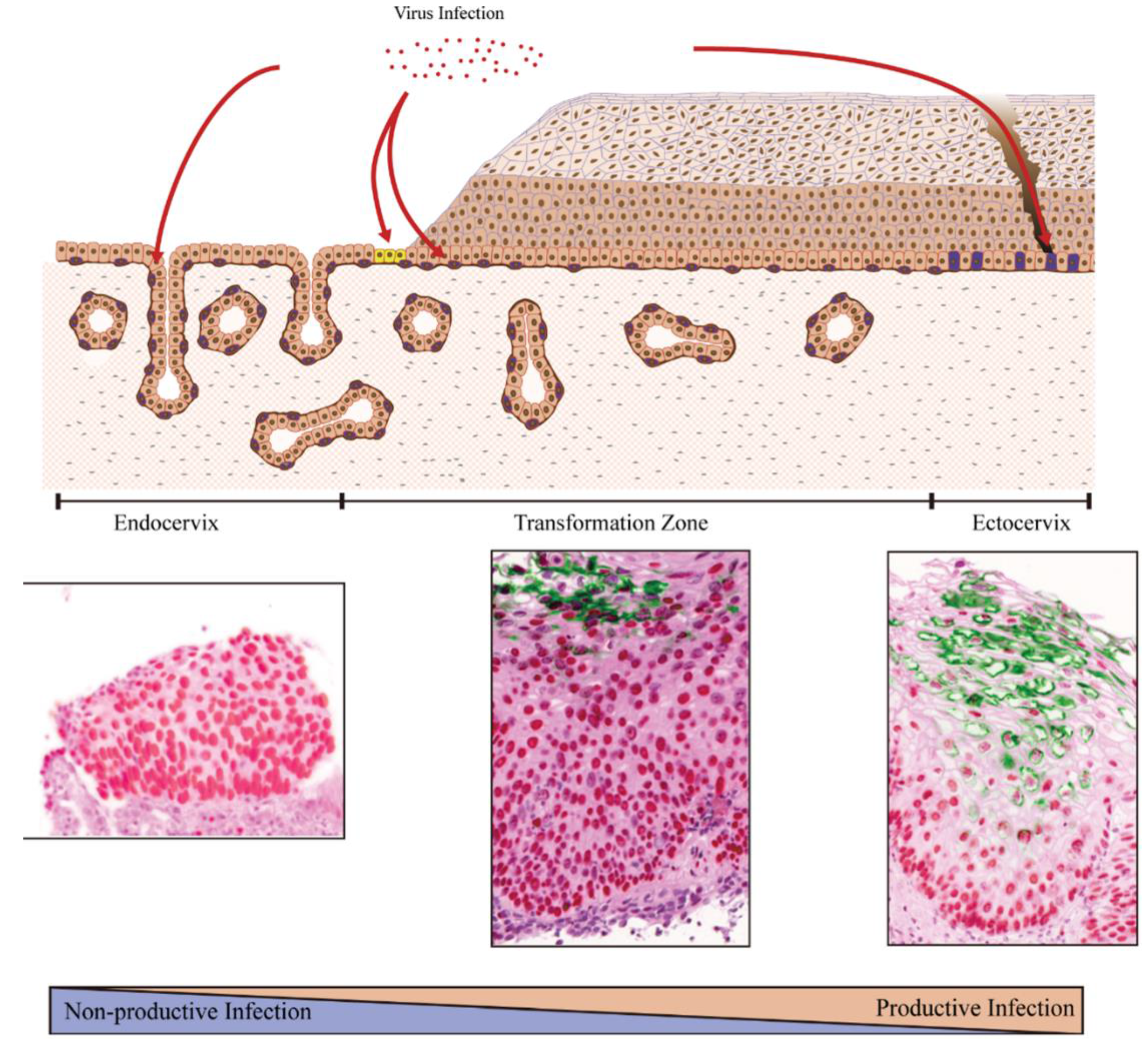
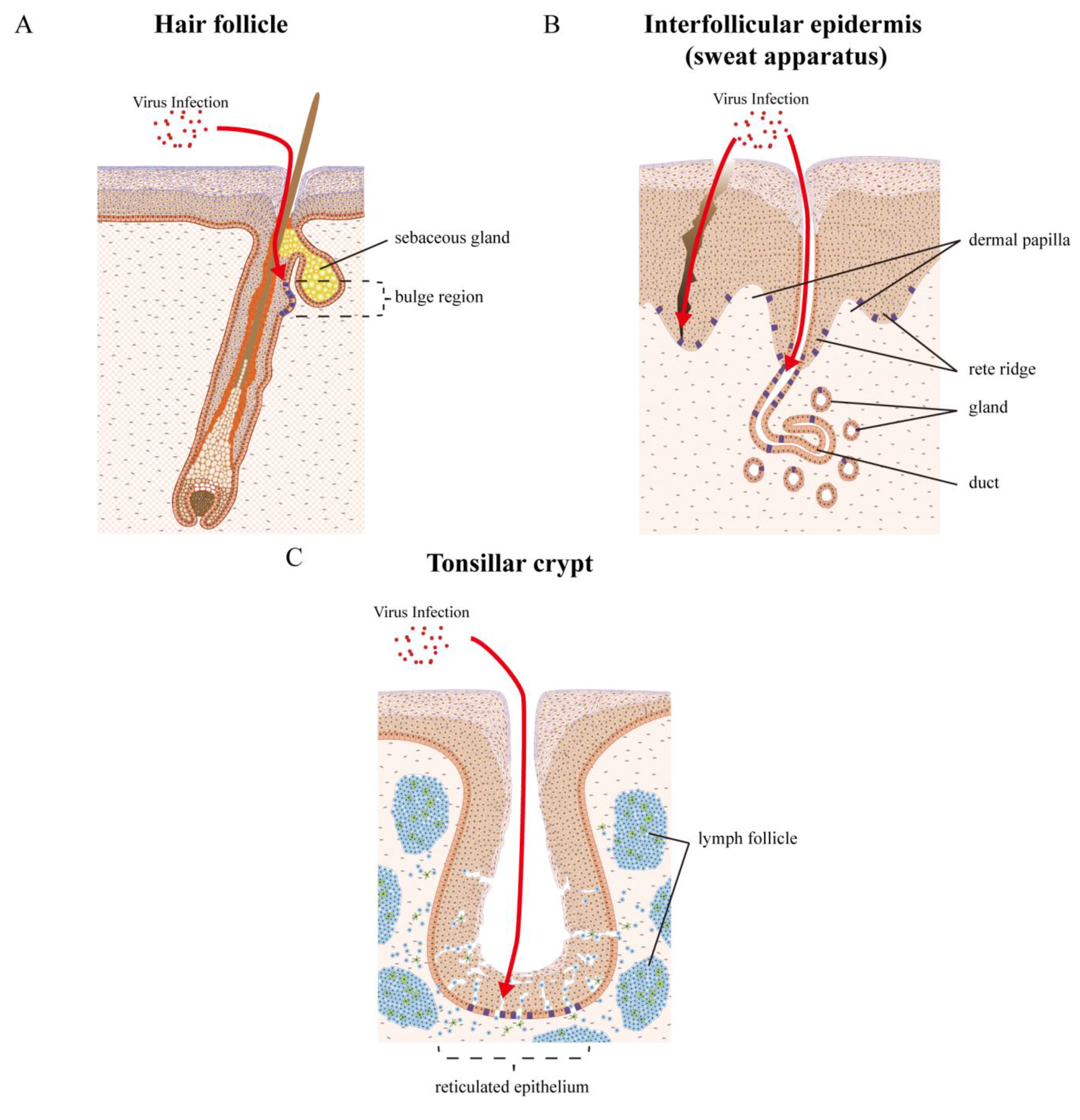
6. Local Epithelial Structure and Sites of Papillomavirus Infection
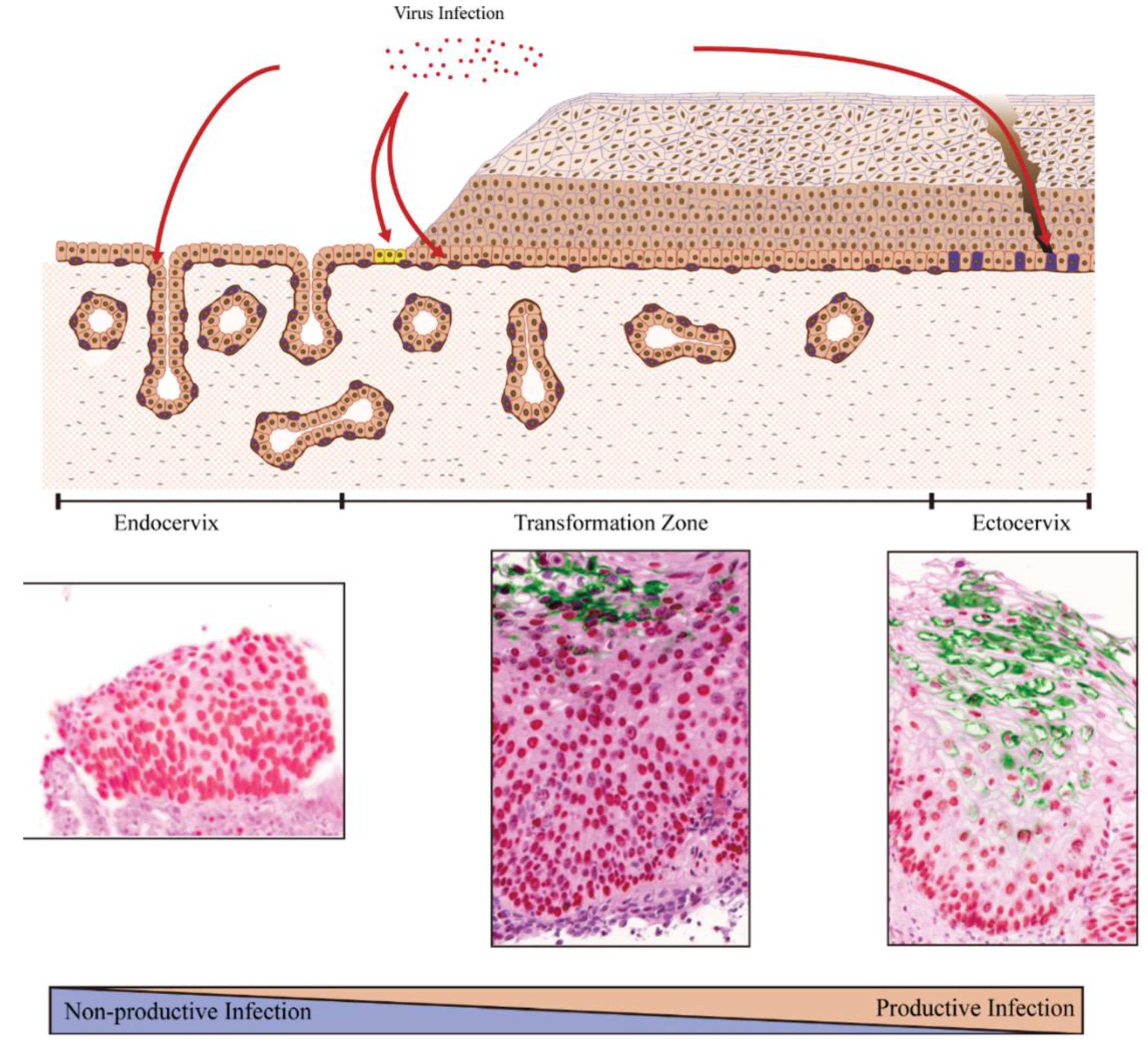
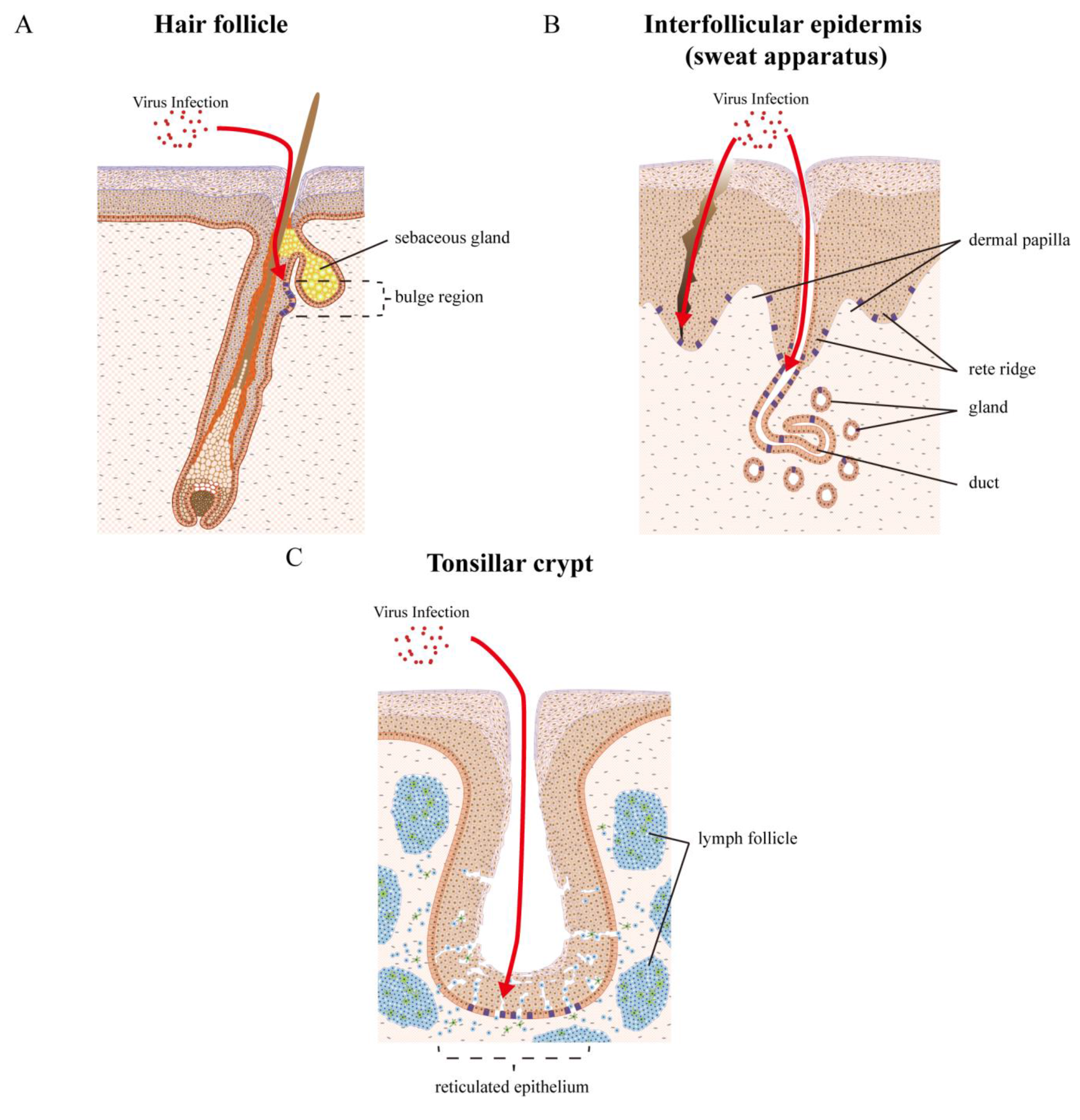
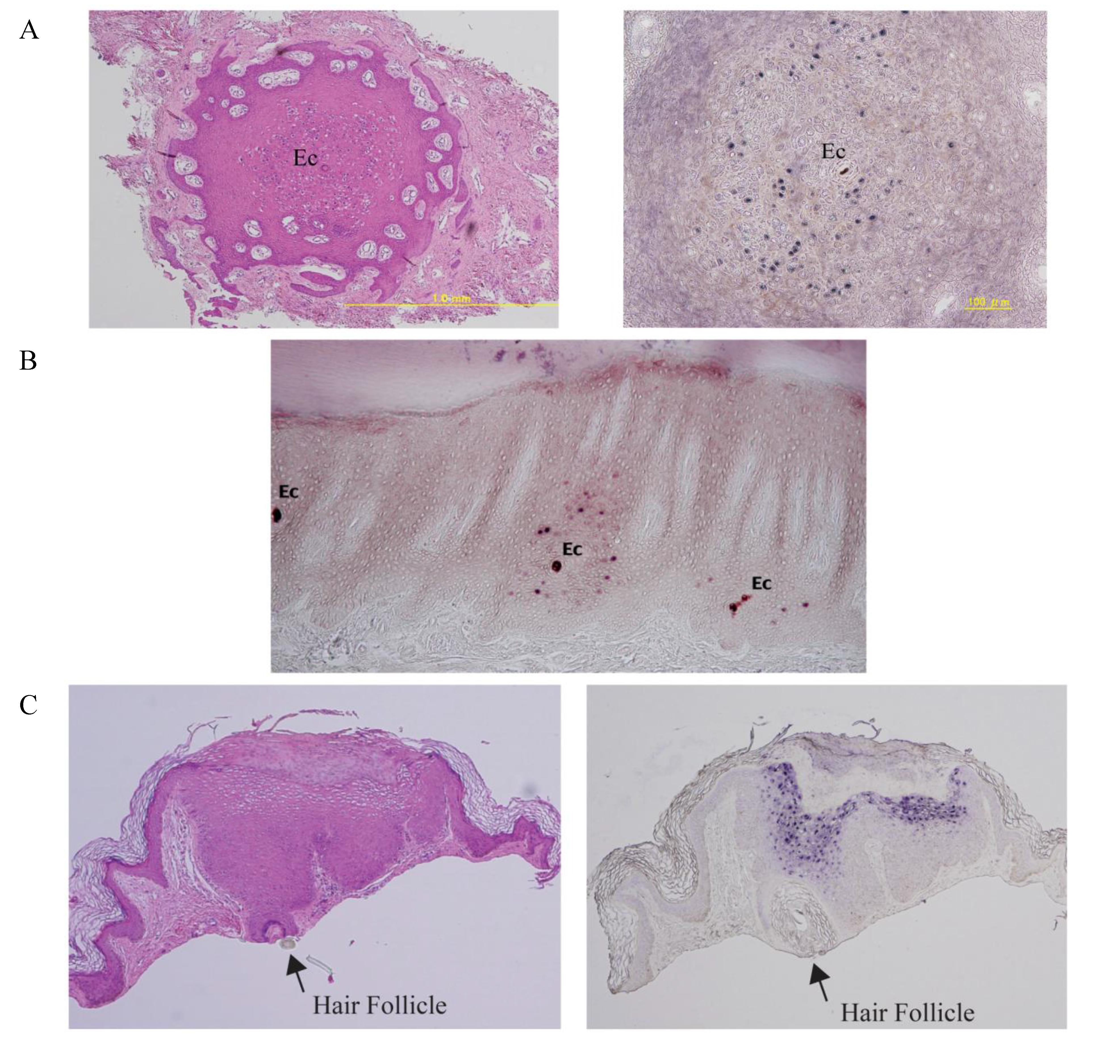
7. Examples of Papillomavirus Niche-Adaptation and Tropisms

8. High-Risk Mucosal HPV Types
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- PaVE: Papillomavirus Episteme. Available online: http://pave.niaid.nih.gov/ (accessed on 11 July 2015).
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Bottalico, D.; Chen, Z.; Dunne, A.; Ostoloza, J.; McKinney, S.; Sun, C.; Schlecht, N.F.; Herrero, R.; Fatahzadeh, M.; Schiffman, M.; et al. The oral cavity contains abundant known and novel human papillomaviruses from the betapapillomavirus and gammapapillomavirus genera. J. Infect. Dis. 2011, 204, 787–792. [Google Scholar] [CrossRef] [PubMed]
- De Koning, M.N.; Quint, K.D.; Bruggink, S.C.; Gussekloo, J.; Bouwes Bavinck, J.N.; Quint, W.G.; Feltkamp, M.C.; Eekhof, J.A. High prevalence of cutaneous warts in elementary school children and the ubiquitous presence of wart-associated human papillomavirus on clinically normal skin. Br. J. Dermatol. 2015, 172, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Poljak, M.; Kocjan, B.J.; Potocnik, M.; Seme, K. Anogenital hairs are an important reservoir of α-papillomaviruses in patients with genital warts. J. Infect. Dis. 2009, 199, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Potocnik, M.; Kocjan, B.J.; Seme, K.; Luzar, B.; Babic, D.Z.; Poljak, M. β-papillomaviruses in anogenital hairs plucked from healthy individuals. J. Med. Virol. 2006, 78, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- De Koning, M.N.; Struijk, L.; Bavinck, J.N.; Kleter, B.; ter Schegget, J.; Quint, W.G.; Feltkamp, M.C. Betapapillomaviruses frequently persist in the skin of healthy individuals. J. Gen. Virol. 2007, 88, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Erfurt, C.; Hazard, K.; Holmgren, V.; Simon, M.; Kataoka, A.; Hossain, S.; Hakangard, C.; Hansson, B.G. Prevalence and type spectrum of human papillomaviruses in healthy skin samples collected in three continents. J. Gen. Virol. 2003, 84, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Karanfilovska, S.; Lindqvist, P.G.; Hansson, B.G. General acquisition of human papillomavirus infections of skin occurs in early infancy. J. Clin. Microbiol. 2003, 41, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, S.; Orth, G. Epidermodysplasia verruciformis. Clin. Dermatol. 1985, 3, 83–96. [Google Scholar] [CrossRef]
- Quint, K.D.; Genders, R.E.; de Koning, M.N.; Borgogna, C.; Gariglio, M.; Bouwes Bavinck, J.N.; Doorbar, J.; Feltkamp, M.C. Human β-papillomavirus infection and keratinocyte carcinomas. J. Pathol. 2015, 235, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.; Sonnex, C.; Jani, P.; Stanley, M.; Sudhoff, H. Recurrent respiratory papillomatosis: An overview of current thinking and treatment. Eur. Arch. Oto-Rhino-Laryngol. 2008, 265, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Cubie, H.A. Diseases associated with human papillomavirus infection. Virology 2013, 445, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Spink, P.J.; Surentheran, T.; Leigh, I.M.; de Villiers, E.M.; McGregor, J.M.; Proby, C.M.; Breuer, J. Degenerate and nested PCR: A highly sensitive and specific method for detection of human papillomavirus infection in cutaneous warts. J. Clin. Microbiol. 1999, 37, 3545–3555. [Google Scholar] [PubMed]
- Rogers, H.D.; Macgregor, J.L.; Nord, K.M.; Tyring, S.; Rady, P.; Engler, D.E.; Grossman, M.E. Acquired epidermodysplasia verruciformis. J. Am. Acad. Dermatol. 2009, 60, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Bouwes Bavinck, J.N.; Feltkamp, M.; Struijk, L.; ter Schegget, J. Human papillomavirus infection and skin cancer risk in organ transplant recipients. J. Investig. Dermatol. Symp. Proc. Soc. 2001, 6, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.G.; Felez-Sanchez, M. Papillomaviruses: Viral evolution, cancer and evolutionary medicine. Evol. Med. Public Health 2015, 2015, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Burk, R.D. Evolution of human papillomavirus carcinogenicity. Adv. Virus Res. 2010, 77, 41–62. [Google Scholar] [PubMed]
- Burk, R.D.; Chen, Z.; van Doorslaer, K. Human papillomaviruses: Genetic basis of carcinogenicity. Public Health Genomics 2009, 12, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVS) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Drolet, M.; Benard, E.; Boily, M.C.; Ali, H.; Baandrup, L.; Bauer, H.; Beddows, S.; Brisson, J.; Brotherton, J.M.; Cummings, T.; et al. Population-level impact and herd effects following human papillomavirus vaccination programmes: A systematic review and meta-analysis. Lancet. Infect. Dis. 2015, 15, 565–580. [Google Scholar] [CrossRef]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, J.; Bienkowska-Haba, M.; Ortega, M.E.; Patel, H.D.; Bodevin, S.; Spillmann, D.; Bishop, B.; Sapp, M.; Chen, X.S. Structural basis of oligosaccharide receptor recognition by human papillomavirus. J. Biol. Chem. 2011, 286, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K. New types of human papillomaviruses and intracytoplasmic inclusion bodies: A classification of inclusion warts according to clinical features, histology and associated HPV types. Br. J. Dermatol. 1994, 130, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K.; Honda, Y.; Inaba, Y.; Ono, T. Pigmented viral warts: A clinical and histopathological study including human papillomavirus typing. Br. J. Dermatol. 1998, 138, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K.; Delius, H.; Matsukura, T.; Kawashima, M.; de Villiers, E.M. Two novel types of human papillomavirus, HPV 63 and HPV 65: Comparisons of their clinical and histological features and DNA sequences to other HPV types. Virology 1993, 194, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K.; Kimmel, R.; de Villiers, E.M. A novel type of human papillomavirus (HPV 95): Comparison with infections of closely related human papillomavirus types. Br. J. Dermatol. 2005, 153, 688–689. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.L.; Winder, D.M.; Vaughan, K.; Hanna, N.; Levy, J.; Sterling, J.C.; Stanley, M.A.; Goon, P.K. Analyses of human papillomavirus genotypes and viral loads in anogenital warts. J. Med. Virol. 2011, 83, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Dickens, P.; Srivastava, G.; Loke, S.L.; Larkin, S. Human papillomavirus 6, 11, and 16 in laryngeal papillomas. J. Pathol. 1991, 165, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S. Human papillomavirus infections and oral tumors. Med. Microbiol. Immunol. Berl 2003, 192, 123–128. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M. Human pathogenic papillomavirus types: An update. Curr. Top. Microbiol. Immunol. 1994, 186, 1–12. [Google Scholar] [PubMed]
- Orth, G.; Jablonska, S.; Favre, M.; Croissant, O.; Obalek, S.; Jarzabek-Chorzelska, M.; Jibard, N. Identification of papillomaviruses in butchers’ warts. J. Investig. Dermatol. 1981, 76, 97–102. [Google Scholar] [CrossRef] [PubMed]
- International Agency for the Research on Cancer. Avilable online: http://monographs.iarc.fr/ENG/Monographs/vol90/ (acessed on 11 July 2015).
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J.; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Herrero, R.; Castellsague, X.; Pawlita, M.; Lissowska, J.; Kee, F.; Balaram, P.; Rajkumar, T.; Sridhar, H.; Rose, B.; Pintos, J.; et al. Human papillomavirus and oral cancer: The international agency for research on cancer multicenter study. J. Natl. Cancer Inst. 2003, 95, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Schelhaas, M. Concepts of papillomavirus entry into host cells. Curr. Opin. Virol. 2014, 4, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.M.; Auborn, K.J.; Brandsma, J.L.; Taichman, L.B. Tissue site-specific enhancer function of the upstream regulatory region of human papillomavirus type 11 in cultured keratinocytes. J. Virol. 1989, 63, 957–960. [Google Scholar] [PubMed]
- Ottinger, M.; Smith, J.A.; Schweiger, M.R.; Robbins, D.; Powell, M.L.; You, J.; Howley, P.M. Cell-type specific transcriptional activities among different papillomavirus long control regions and their regulation by E2. Virology 2009, 395, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Mistry, N.; Wibom, C.; Evander, M. Cutaneous and mucosal human papillomaviruses differ in net surface charge, potential impact on tropism. Virol. J. 2008, 5. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K.; Inaba, Y.; Yoshimura, K.; Ono, T. Varied clinical morphology of HPV-1-induced warts, depending on anatomical factors. Br. J. Dermatol. 1993, 128, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Trenfield, K.; Spradbrow, P.B.; Vanselow, B. Sequences of papillomavirus DNA in equine sarcoids. Equine Vet. J. 1985, 17, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Otten, N.; von Tscharner, C.; Lazary, S.; Antczak, D.F.; Gerber, H. DNA of bovine papillomavirus type 1 and 2 in equine sarcoids: Pcr detection and direct sequencing. Arch. Virol. 1993, 132, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Angelos, J.A.; Marti, E.; Lazary, S.; Carmichael, L.E. Characterization of BPV-like DNA in equine sarcoids. Arch. Virol. 1991, 119, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Chambers, G.; Ellsmore, V.A.; O’Brien, P.M.; Reid, S.W.; Love, S.; Campo, M.S.; Nasir, L. Association of bovine papillomavirus with the equine sarcoid. J. Gen. Virol. 2003, 84, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Rous, P.; Beard, J.W. The progression to carcinoma of virus-induced rabbit papillomas (shope). J. Exp. Med. 1935, 62, 523–548. [Google Scholar] [CrossRef] [PubMed]
- Syverton, J.T. The pathogenesis of the rabbit papilloma-to-carcinoma sequence. Ann. N. Y. Acad. Sci. 1952, 54, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Jeckel, S.; Huber, E.; Stubenrauch, F.; Iftner, T. A transactivator function of cottontail rabbit papillomavirus E2 is essential for tumor induction in rabbits. J. Virol. 2002, 76, 11209–11215. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Harry, J.; Lin, Y.L.; Wettstein, F.O. Identification of three transforming proteins encoded by cottontail rabbit papillomavirus. J. Virol. 1992, 66, 1655–1664. [Google Scholar] [PubMed]
- Gjoerup, O.; Chang, Y. Update on human polyomaviruses and cancer. Adv. Cancer Res. 2010, 106, 1–51. [Google Scholar] [PubMed]
- Schlesinger, R.W. Adenoviruses: The nature of the virion and of controlling factors in productive or abortive infection and tumorigenesis. Adv. Virus Res. 1969, 14, 1–61. [Google Scholar] [PubMed]
- Pipas, J.M. SV40: Cell transformation and tumorigenesis. Virology 2009, 384, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Endter, C.; Dobner, T. Cell transformation by human adenoviruses. Curr. Top. Microbiol. Immunol. 2004, 273, 163–214. [Google Scholar] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Middleton, K.; Peh, W.; Southern, S.; Griffin, H.; Sotlar, K.; Nakahara, T.; El-Sherif, A.; Morris, L.; Seth, R.; Hibma, M.; et al. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J. Virol. 2003, 77, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.; Soneji, Y.; van Baars, R.; Arora, R.; Jenkins, D.; van de Sandt, M.; Wu, Z.; Quint, W.; Jach, R.; Okon, K.; et al. Stratification of HPV-induced cervical pathology using the virally encoded molecular marker E4 in conbination with p16 or mcm. Mod. Pathol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Pater, M.M.; Tang, S.C.; Pater, A. Effect of glucocorticoid hormones on viral gene expression, growth, and dysplastic differentiation in HPV16-immortalized ectocervical cells. Exp. Cell Res. 1997, 232, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Tsutsumi, K.; Pater, A.; Pater, M.M. Human papillomavirus type 16 expression in cervical keratinocytes: Role of progesterone and glucocorticoid hormones. Obstet. Gynecol. 1993, 81, 5–12. [Google Scholar] [PubMed]
- Kyo, S.; Inoue, M.; Hayasaka, N.; Inoue, T.; Yutsudo, M.; Tanizawa, O.; Hakura, A. Regulation of early gene expression of human papillomavirus type 16 by inflammatory cytokines. Virology 1994, 200, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Forslund, O.; Ekberg, H.; Sterner, G.; Hansson, B.G. The ubiquity and impressive genomic diversity of human skin papillomaviruses suggest a commensalic nature of these viruses. J. Virol. 2000, 74, 11636–11641. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Hansson, B.G. Healthy skin of many animal species harbors papillomaviruses which are closely related to their human counterparts. J. Virol. 2002, 76, 12537–12542. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.B.; Doorbar, J. Immunosuppression facilitates the reactivation of latent papillomavirus infections. J. Virol. 2014, 88, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Latent papillomavirus infections and their regulation. Curr. Opin. Virol. 2013, 3, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E. Evidence and impact of human papillomavirus latency. Open Virol. J. 2012, 6, 198–203. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Delius, H.; van Ranst, M.A.; Jenson, A.B.; zur Hausen, H.; Sundberg, J.P. Canine oral papillomavirus genomic sequence: A unique 1.5-kb intervening sequence between the E2 and L2 open reading frames. Virology 1994, 204, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Terai, M.; Burk, R.D. Felis domesticus papillomavirus, isolated from a skin lesion, is related to canine oral papillomavirus and contains a 1.3 kb non-coding region between the E2 and L2 open reading frames. J. Gen. Virol. 2002, 83, 2303–2307. [Google Scholar] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Holmgren, S.C.; Patterson, N.A.; Ozbun, M.A.; Lambert, P.F. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J. Virol. 2005, 79, 3938–3948. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. Chlr1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. Lond. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Melendy, T.; Sedman, J.; Stenlund, A. Cellular factors required for papillomavirus DNA replication. J. Virol. 1995, 69, 7857–7867. [Google Scholar] [PubMed]
- White, E.A.; Howley, P.M. Proteomic approaches to the study of papillomavirus-host interactions. Virology 2013, 435, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Lin, B.Y.; Deng, W.; Broker, T.R.; Chow, L.T. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J. Virol. 2007, 81, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Moody, C.; Laimins, L.A.; Archambault, J. Nuclear export of human papillomavirus type 31 E1 is regulated by CDK2 phosphorylation and required for viral genome maintenance. J. Virol. 2010, 84, 11747–11760. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.B.; Laskey, P.; Sullivan, K.; Davy, C.; Wang, Q.; Jackson, D.J.; Griffin, H.M.; Doorbar, J. E1–E4-mediated keratin phosphorylation and ubiquitylation: A mechanism for keratin depletion in HPV16-infected epithelium. J. Cell Sci. 2010, 123, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.B.; Martin, S.R.; Jackson, D.J.; Khan, J.; Isaacson, E.R.; Calder, L.; Raj, K.; Griffin, H.M.; Wang, Q.; Laskey, P.; et al. Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1–E4 protein and provides a molecular basis for its accumulation. J. Virol. 2008, 82, 8196–8203. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P.; Klaunberg, B.; Moore, R.A.; Santos, E.B.; Parry, N.R.; Gough, G.W.; Stanley, M.A. Naturally occurring, nonregressing canine oral papillomavirus infection: Host immunity, virus characterization, and experimental infection. Virology 1999, 265, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Wilgenburg, B.J.; Budgeon, L.R.; Lang, C.M.; Griffith, J.W.; Christensen, N.D. Characterization of immune responses during regression of rabbit oral papillomavirus infections. Comp. Med. 2005, 55, 431–439. [Google Scholar] [PubMed]
- Nicholls, P.K.; Moore, P.F.; Anderson, D.M.; Moore, R.A.; Parry, N.R.; Gough, G.W.; Stanley, M.A. Regression of canine oral papillomas is associated with infiltration of CD4+ and CD8+ lymphocytes. Virology 2001, 283, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Birley, H.D.; Renton, A.M.; Hanna, N.F.; Ryait, B.K.; Byrne, M.; Taylor-Robinson, D.; Stanley, M.A. Immunological events in regressing genital warts. Am. J. Clin. Pathol. 1994, 102, 768–774. [Google Scholar] [PubMed]
- Monnier-Benoit, S.; Mauny, F.; Riethmuller, D.; Guerrini, J.S.; Capilna, M.; Felix, S.; Seilles, E.; Mougin, C.; Pretet, J.L. Immunohistochemical analysis of CD4+ and CD8+ T-cell subsets in high risk human papillomavirus-associated pre-malignant and malignant lesions of the uterine cervix. Gynecol. Oncol. 2006, 102, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kanodia, S.; Fahey, L.M.; Kast, W.M. Mechanisms used by human papillomaviruses to escape the host immune response. Curr. Cancer Drug Targets 2007, 7, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K. Do human papillomaviruses target epidermal stem cells? Dermatology 2003, 207, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Vargas, S.O.; Yamamoto, Y.; Howitt, B.E.; Nucci, M.R.; Hornick, J.L.; McKeon, F.D.; Xian, W.; Crum, C.P. A novel blueprint for “top down” differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. J. Pathol. 2013, 229, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Yamamoto, Y.; Laury, A.; Wang, X.; Nucci, M.R.; McLaughlin-Drubin, M.E.; Munger, K.; Feldman, S.; McKeon, F.D.; Xian, W.; et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M. Epidermal stem cells: Markers, patterning and the control of stem cell fate. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1998, 353, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S. Cell replacement in epidermis (keratopoiesis) via discrete units of proliferation. Int. Rev. Cytol. 1981, 69, 271–318. [Google Scholar] [PubMed]
- Feuerman, E. Verruca vulgaris—Benign tumor of viral etiology and clonal development. Harefuah 1972, 83, 211–212. [Google Scholar] [PubMed]
- Schmitt, A.; Rochat, A.; Zeltner, R.; Borenstein, L.; Barrandon, Y.; Wettstein, F.O.; Iftner, T. The primary target cells of the high-risk cottontail rabbit papillomavirus colocalize with hair follicle stem cells. J. Virol. 1996, 70, 1912–1922. [Google Scholar] [PubMed]
- Martens, J.E.; Smedts, F.M.; Ploeger, D.; Helmerhorst, T.J.; Ramaekers, F.C.; Arends, J.W.; Hopman, A.H. Distribution pattern and marker profile show two subpopulations of reserve cells in the endocervical canal. Int. J. Gynecol. Pathol. 2009, 28, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, J.; Howitt, B.E.; Roncarati, P.; Demoulin, S.; Suarez-Carmona, M.; Hubert, P.; McKeon, F.D.; Xian, W.; Li, A.; Delvenne, P.; et al. Carcinogenic HPV infection in the cervical squamo-columnar junction. J. Pathol. 2015, 236, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Isaacson Wechsler, E.; Wang, Q.; Roberts, I.; Pagliarulo, E.; Jackson, D.; Untersperger, C.; Coleman, N.; Griffin, H.; Doorbar, J. Reconstruction of human papillomavirus type 16-mediated early-stage neoplasia implicates E6/E7 deregulation and the loss of contact inhibition in neoplastic progression. J. Virol. 2012, 86, 6358–6364. [Google Scholar] [CrossRef] [PubMed]
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar] [CrossRef]
- Cotsarelis, G. Epithelial stem cells: A folliculocentric view. J. Investig. Dermatol. 2006, 126, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Organization of stem cells and their progeny in human epidermis. J. Investig. Dermatol. 2005, 124, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.; Lehrer, M.S.; Jensen, P.J.; Sun, T.T.; Lavker, R.M. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell 2000, 102, 451–461. [Google Scholar] [CrossRef]
- Ito, M.; Liu, Y.; Yang, Z.; Nguyen, J.; Liang, F.; Morris, R.J.; Cotsarelis, G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 2005, 11, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P. Interfollicular epidermal stem cells: Identification, challenges, potential. J. Investig. Dermatol. 2006, 126, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.M.; Niemann, C.; Jensen, U.B.; Sundberg, J.P.; Silva-Vargas, V.; Watt, F.M. Manipulation of stem cell proliferation and lineage commitment: Visualisation of label-retaining cells in wholemounts of mouse epidermis. Development 2003, 130, 5241–5255. [Google Scholar] [CrossRef] [PubMed]
- Lavker, R.M.; Sun, T.T. Epidermal stem cells. J. Investig. Dermatol. 1983, 81, 121s–127s. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Multiple classes of stem cells in cutaneous epithelium: A lineage analysis of adult mouse skin. EMBO J. 2001, 20, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J.; Burke, E.M.; Rader, M.D.; Coulombe, P.A.; Lavker, R.M. Re-epithelialization of porcine skin by the sweat apparatus. J. Investig. Dermatol. 1998, 110, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.P.; Polak, L.; Rocha, A.S.; Pasolli, H.A.; Chen, S.C.; Sharma, N.; Blanpain, C.; Fuchs, E. Identification of stem cell populations in sweat glands and ducts reveals roles in homeostasis and wound repair. Cell 2012, 150, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, S.; Dabrowski, J.; Jakubowicz, K. Epidermodysplasia verruciformis as a model in studies on the role of papovaviruses in oncogenesis. Cancer Res. 1972, 32, 583–589. [Google Scholar] [PubMed]
- Orth, G. Genetics of epidermodysplasia verruciformis: Insights into host defense against papillomaviruses. Semin. Immunol. 2006, 18, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Lazarczyk, M.; Dalard, C.; Hayder, M.; Dupre, L.; Pignolet, B.; Majewski, S.; Vuillier, F.; Favre, M.; Liblau, R.S. Ever proteins, key elements of the natural anti-human papillomavirus barrier, are regulated upon t-cell activation. PLoS ONE 2012, 7, e39995. [Google Scholar] [CrossRef] [PubMed]
- Boxman, I.L.; Berkhout, R.J.; Mulder, L.H.; Wolkers, M.C.; Bouwes Bavinck, J.N.; Vermeer, B.J.; ter Schegget, J. Detection of human papillomavirus DNA in plucked hairs from renal transplant recipients and healthy volunteers. J. Investig. Dermatol. 1997, 108, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Boxman, I.L.; Mulder, L.H.; Russell, A.; Bouwes Bavinck, J.N.; Green, A.; Ter Schegget, J. Human papillomavirus type 5 is commonly present in immunosuppressed and immunocompetent individuals. Br. J. Dermatol. 1999, 141, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, S.; Orth, G.; Obalek, S.; Croissant, O. Cutaneous warts. Clinical, histologic, and virologic correlations. Clin. Dermatol. 1985, 3, 71–82. [Google Scholar] [CrossRef]
- Egawa, K. Eccrine-centred distribution of human papillomavirus 63 infection in the epidermis of the plantar skin. Br. J. Dermatol. 2005, 152, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Boxman, I.L.; Hogewoning, A.; Mulder, L.H.; Bouwes Bavinck, J.N.; ter Schegget, J. Detection of human papillomavirus types 6 and 11 in pubic and perianal hair from patients with genital warts. J. Clin. Microbiol. 1999, 37, 2270–2273. [Google Scholar] [PubMed]
- Kocjan, B.J.; Poljak, M.; Seme, K.; Potocnik, M.; Fujs, K.; Babic, D.Z. Distribution of human papillomavirus genotypes in plucked eyebrow hairs from slovenian males with genital warts. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2005, 5, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T. Cell polarity proteins: Common targets for tumorigenic human viruses. Oncogene 2008, 27, 7031–7046. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Banks, L. A systematic analysis of human papillomavirus (HPV) E6 pdz substrates identifies magi-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J. Virol. 2011, 85, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Narayan, N.; Thomas, M.; Gammoh, N.; Strand, S.; Strand, D.; Banks, L. Regulation of the hdlg/hscrib/hugl-1 tumour suppressor complex. Exp. Cell Res. 2008, 314, 3306–3317. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaic, V.; Banks, L. Human papillomaviruses and the specificity of pdz domain targeting. FEBS J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Dasgupta, J.; Zhang, Y.; Chen, X.; Banks, L. Analysis of specificity determinants in the interactions of different HPV E6 proteins with their pdz domain-containing substrates. Virology 2008, 376, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; van Doorslaer, K.; Chen, Z.; Ristriani, T.; Masson, M.; Trave, G.; Burk, R.D. Degradation of p53 by human alphapapillomavirus E6 proteins shows a stronger correlation with phylogeny than oncogenicity. PLoS ONE 2010, 5, e12816. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; ould M’hamed ould Sidi, A.; Boulade-Ladame, C.; Rybin, V.; Chappelle, A.; Atkinson, A.; Kieffer, B.; Trave, G. Solution structure analysis of the HPV16 E6 oncoprotein reveals a self-association mechanism required for E6-mediated degradation of p53. Structure 2012, 20, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Banks, L. Interaction of viral oncoproteins with cellular target molecules: Infection with high-risk vs. low-risk human papillomaviruses. Apmis 2010, 118, 471–493. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Baker, C.C. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Westra, W.H. The morphologic profile of HPV-related head and neck squamous carcinoma: Implications for diagnosis, prognosis, and clinical management. Head Neck Pathol. 2012, 6, S48–S54. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer J. Int. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; Castellsague, X.; Diaz, M.; de Sanjose, S.; Hammouda, D.; Shah, K.V.; Meijer, C.J. Against which human papillomavirus types shall we vaccinate and screen? The international perspective. Int. J. Cancer J. Int. Cancer 2004, 111, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Hagedorn, M.; Ikenberg, H.; Rufli, T.; Dahlet, C.; Grosshans, E.; Gissmann, L. Bowenoid papulosis. Presence of human papillomavirus (HPV) structural antigens and of HPV 16-related DNA sequences. Arch. Dermatol. 1985, 121, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Tschandl, P.; Rosendahl, C.; Kittler, H. Cutaneous human papillomavirus infection: Manifestations and diagnosis. Curr. Probl. Dermatol. 2014, 45, 92–97. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863-3890. https://doi.org/10.3390/v7072802
Egawa N, Egawa K, Griffin H, Doorbar J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses. 2015; 7(7):3863-3890. https://doi.org/10.3390/v7072802
Chicago/Turabian StyleEgawa, Nagayasu, Kiyofumi Egawa, Heather Griffin, and John Doorbar. 2015. "Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia" Viruses 7, no. 7: 3863-3890. https://doi.org/10.3390/v7072802
APA StyleEgawa, N., Egawa, K., Griffin, H., & Doorbar, J. (2015). Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses, 7(7), 3863-3890. https://doi.org/10.3390/v7072802