
Coordinated DNA Replication by the Bacteriophage T4 Replisome

{kind=link}
{kind=link}
Abstract
:1. Introduction
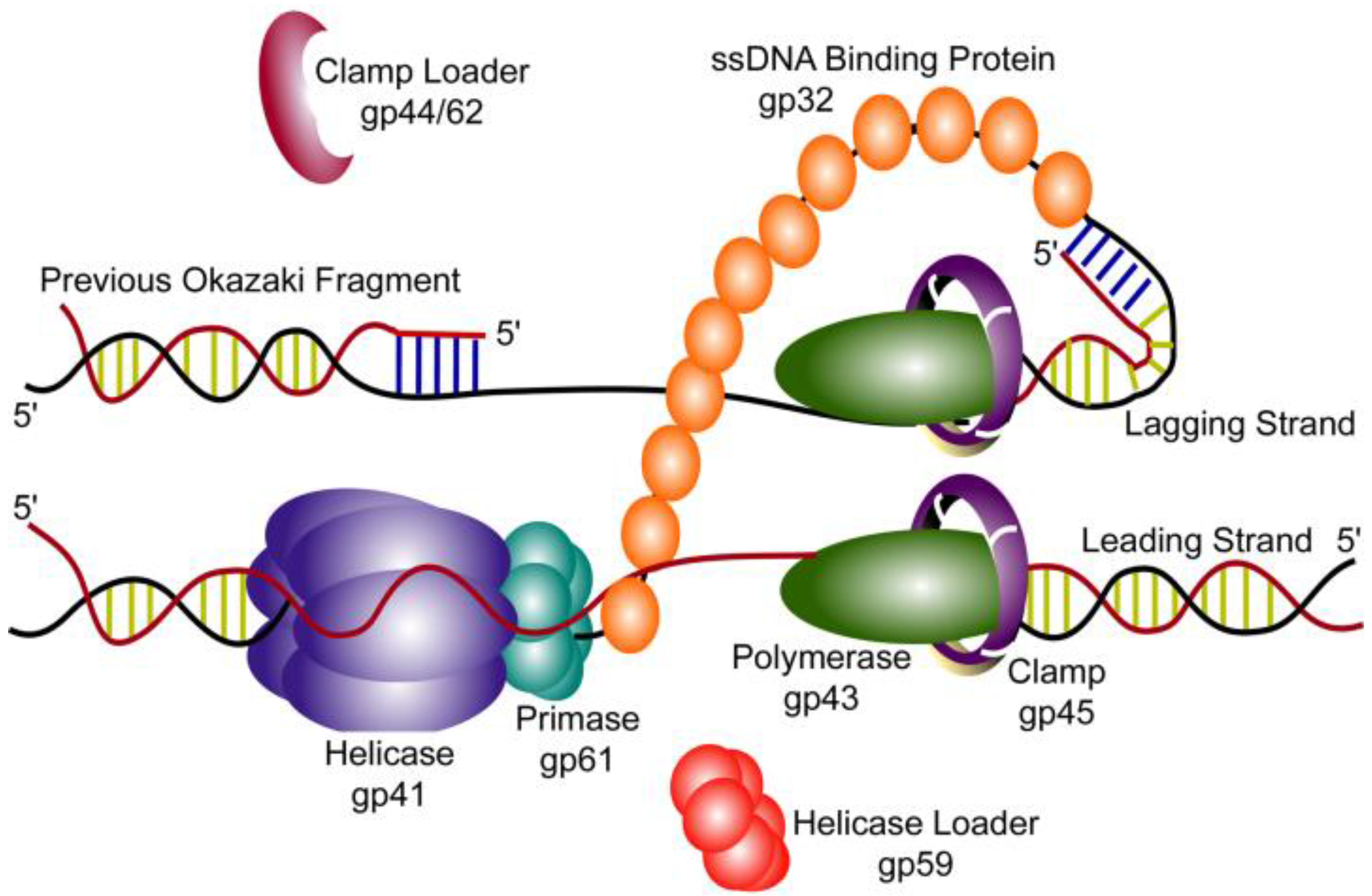
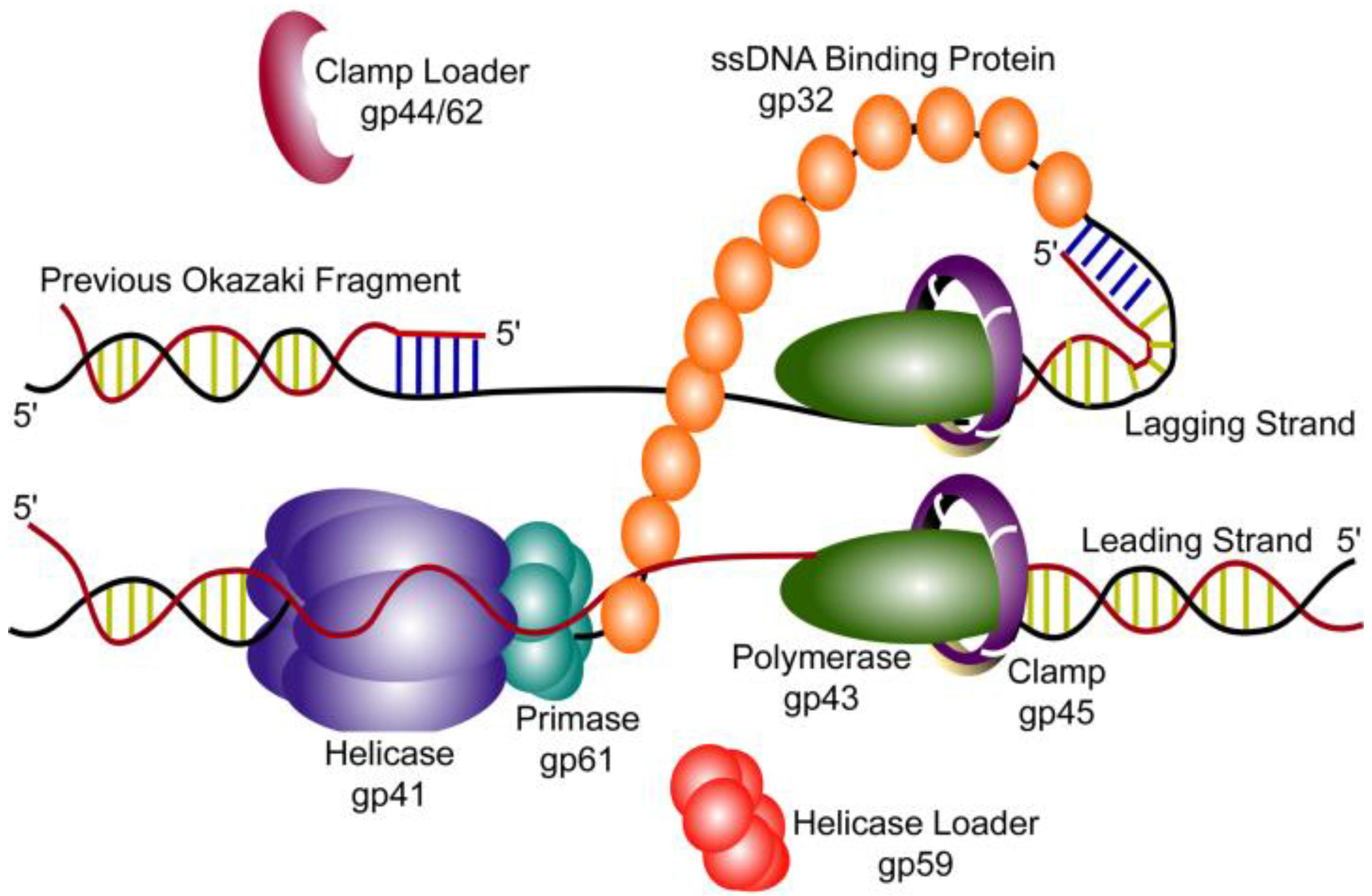
2. T4 Replication Fork Components
3. Holoenzyme Formation
4. Holoenzyme Processivity
5. Coupling of Helicase and Polymerase for Leading Strand Synthesis
6. Coordination of Helicase and Priming on the Lagging Strand
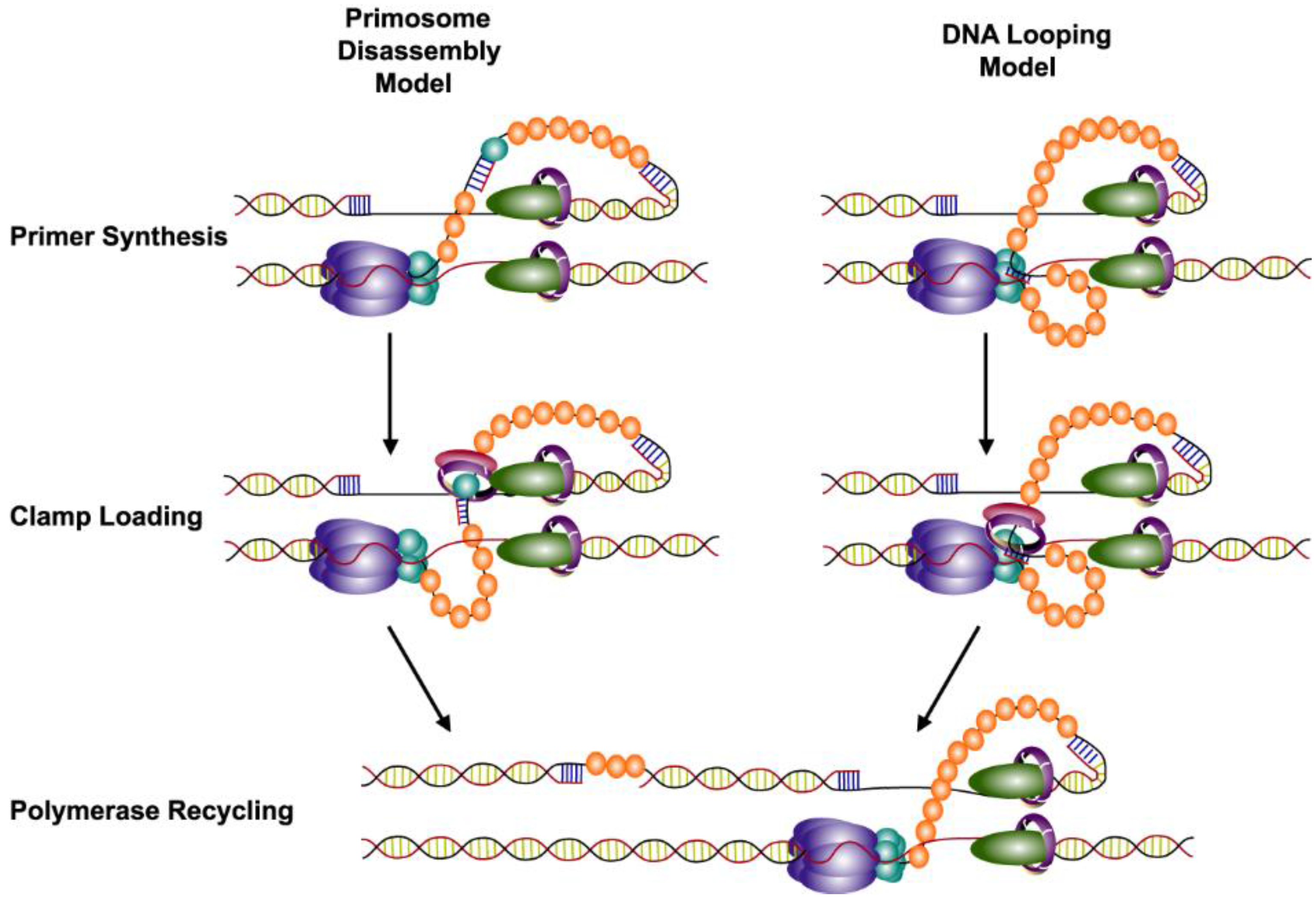
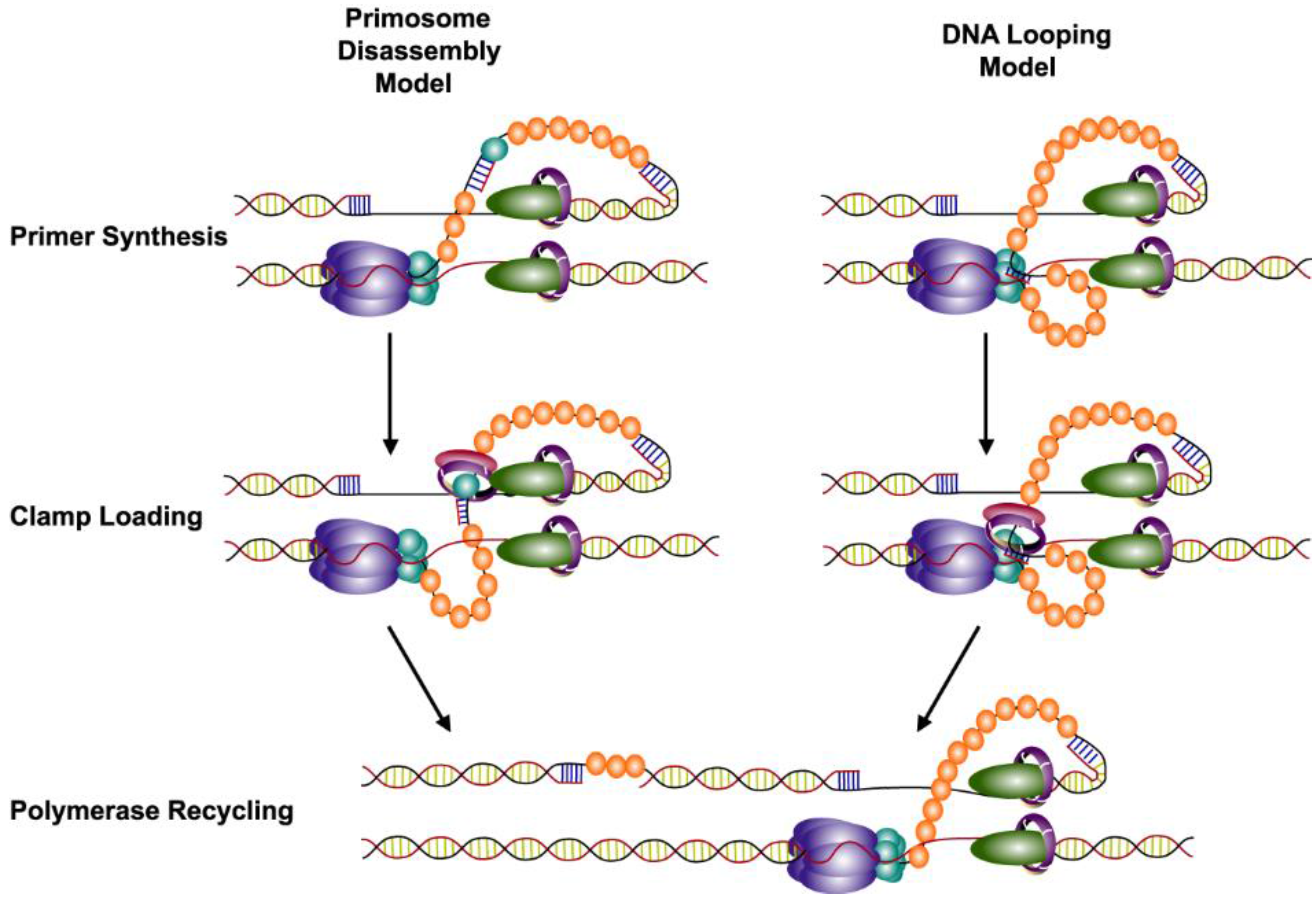
7. Recycling of the Lagging Strand Polymerase
8. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Nossal, N.G. Protein-protein interactions at a DNA replication fork: Bacteriophage T4 as a model. FASEB J. 1992, 6, 871–878. [Google Scholar] [PubMed]
- Liu, C.; Burke, R.; Hibner, U.; Barry, J.; Alberts, B. Probing DNA Replication Mechanisms with the T4 Bacteriophage in Vitro System. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1979; pp. 469–487. [Google Scholar]
- Alberts, B.; Barry, J.; Bedinger, P.; Formosa, T.; Jongeneel, C.; Kreuzer, K. Studies on DNA Replication in the Bacteriophage T4 in Vitro System. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1983; pp. 655–668. [Google Scholar]
- Mueser, T.C.; Hinerman, J.M.; Devos, J.M.; Boyer, R.A.; Williams, K.J. Structural analysis of bacteriophage T4 DNA replication: A review in the virology journal series on bacteriophage T4 and its relatives. Virol. J. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Richardson, C.C. Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 2011, 15, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Richardson, C.C. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009, 78, 205–243. [Google Scholar] [CrossRef] [PubMed]
- Van Oijen, A.M.; Loparo, J.J. Single-molecule studies of the replisome. Annu. Rev. Biophys. 2010, 39, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Mosig, G.; Colowick, N.; Gruidl, M.E.; Chang, A.; Harvey, A.J. Multiple initiation mechanisms adapt phage T4 DNA replication to physiological changes during T4’s development. FEMS Microb. Rev. 1995, 17, 83–98. [Google Scholar] [CrossRef]
- Kreuzer, K.N.; Brister, J.R. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: A review in the virology journal series on bacteriophage T4 and its relatives. Virol. J. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, S.J.; Valentine, A.M.; Salinas, F. Replisome-mediated DNA replication. Annu. Rev. Biochem. 2001, 70, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.K.; Weitzel, S.E.; von Hippel, P.H. Assembly of a functional replication complex without ATP hydrolysis: A direct interaction of bacteriophage T4 gp45 with T4 DNA polymerase. Proc. Natl. Acad. Sci. USA 1993, 90, 3211–3215. [Google Scholar] [CrossRef] [PubMed]
- Sexton, D.J.; Berdis, A.J.; Benkovic, S.J. Assembly and disassembly of DNA polymerase holoenzyme. Curr. Opin. Chem. Biol. 1997, 1, 316–322. [Google Scholar] [CrossRef]
- Franklin, M.C.; Wang, J.; Steitz, T.A. Structure of the replicating complex of a pol α family DNA polymerase. Cell 2001, 105, 657–667. [Google Scholar] [CrossRef]
- Wang, J.; Sattar, A.A.; Wang, C.; Karam, J.; Konigsberg, W.; Steitz, T. Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell 1997, 89, 1087–1099. [Google Scholar] [CrossRef]
- Shamoo, Y.; Steitz, T.A. Building a replisome from interacting pieces: Sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 1999, 99, 155–166. [Google Scholar] [CrossRef]
- Lin, T.-C.; Karam, G.; Konigsberg, W.H. Isolation, characterization, and kinetic properties of truncated forms of T4 DNA polymerase that exhibit 3′–5′exonuclease activity. J. Biol. Chem. 1994, 269, 19286–19294. [Google Scholar] [PubMed]
- Bruck, I.; O’Donnell, M. The ring-type polymerase sliding clamp family. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Moarefi, I.; Jeruzalmi, D.; Turner, J.; O’Donnell, M.; Kuriyan, J. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 2000, 296, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Soumillion, P.; Sexton, D.J.; Benkovic, S.J. Clamp subunit dissociation dictates bacteriophage T4 DNA polymerase holoenzyme disassembly. Biochemistry 1998, 37, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Shier, V.K.; Abel-Santos, E.; Sexton, D.J.; Soumillion, P.; Benkovic, S.J. Sliding clamp of the bacteriophage T4 polymerase has open and closed subunit interfaces in solution. Biochemistry 1999, 38, 7696–7709. [Google Scholar] [CrossRef] [PubMed]
- Millar, D.; Trakselis, M.A.; Benkovic, S.J. On the solution structure of the T4 sliding clamp (gp45). Biochemistry 2004, 43, 12723–12727. [Google Scholar] [CrossRef] [PubMed]
- Berdis, A.J.; Soumillion, P.; Benkovic, S.J. The carboxyl terminus of the bacteriophage T4 DNA polymerase is required for holoenzyme complex formation. Proc. Natl. Acad. Sci. USA 1996, 93, 12822–12827. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, T.; Paul, L.; Hockensmith, J.; von Hippel, P. Structural and enzymatic studies of the T4 DNA replication system. II. Atpase properties of the polymerase accessory protein complex. J. Biol. Chem. 1989, 264, 12717–12729. [Google Scholar] [PubMed]
- Jarvis, T.; Newport, J.; von Hippel, P. Stimulation of the processivity of the DNA polymerase of bacteriophage T4 by the polymerase accessory proteins. The role of atp hydrolysis. J. Biol. Chem. 1991, 266, 1830–1840. [Google Scholar] [PubMed]
- Rush, J.; Lin, T.; Quinones, M.; Spicer, E.; Douglas, I.; Williams, K.; Konigsberg, W. The 44p subunit of the T4 DNA polymerase accessory protein complex catalyzes ATP hydrolysis. J. Biol. Chem. 1989, 264, 10943–10953. [Google Scholar] [PubMed]
- Bowman, G.D.; O’Donnell, M.; Kuriyan, J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 2004, 429, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, K.R.; Kazmirski, S.L.; Goedken, E.R.; Cantor, A.J.; Kelch, B.A.; McNally, R.; Seyedin, S.N.; Makino, D.L.; O’Donnell, M.; Kuriyan, J. The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 2009, 137, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Gogol, E.P.; von Hippel, P.H. The phage T4-coded DNA replication helicase (gp41) forms a hexamer upon activation by nucleoside triphosphate. J. Biol. Chem. 1995, 270, 7462–7473. [Google Scholar] [PubMed]
- Young, M.C.; Schultz, D.E.; Ring, D.; von Hippel, P.H. Kinetic parameters of the translocation of bacteriophage T4 gene 41 protein helicase on single-stranded DNA. J. Mol. Biol. 1994, 235, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Alberts, B. Characterization of the DNA-dependent gtpase activity of T4 gene 41 protein, an essential component of the t4 bacteriophage DNA replication apparatus. J. Biol. Chem. 1981, 256, 2813–2820. [Google Scholar]
- Norcum, M.T.; Warrington, J.A.; Spiering, M.M.; Ishmael, F.T.; Trakselis, M.A.; Benkovic, S.J. Architecture of the bacteriophage T4 primosome: Electron microscopy studies of helicase (gp41) and primase (gp61). Proc. Natl. Acad. Sci. USA 2005, 102, 3623–3626. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, M.; Silver, L.; Nossal, N. Bacteriophage T4 gene 41 protein, required for the synthesis of RNA primers, is also a DNA helicase. J. Biol. Chem. 1982, 257, 12426–12434. [Google Scholar] [PubMed]
- Richardson, R.W.; Nossal, N. Characterization of the bacteriophage T4 gene 41 DNA helicase. J. Biol. Chem. 1989, 264, 4725–4731. [Google Scholar] [PubMed]
- Perumal, S.K.; Raney, K.D.; Benkovic, S.J. Analysis of the DNA translocation and unwinding activities of T4 phage helicases. Methods 2010, 51, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Spiering, M.M.; Trakselis, M.A.; Ishmael, F.T.; Xi, J.; Benkovic, S.J.; Hammes, G.G. Assembly of the bacteriophage T4 primosome: Single-molecule and ensemble studies. Proc. Natl. Acad. Sci. USA 2005, 102, 3254–3259. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.; Alberts, B. Studies of the DNA helicase-RNA primase unit from bacteriophage T4. A trinucleotide sequence on the DNA template starts rna primer synthesis. J. Biol. Chem. 1986, 261, 7001–7010. [Google Scholar] [PubMed]
- Hinton, D.; Nossal, N. Bacteriophage T4 DNA primase-helicase. Characterization of oligomer synthesis by T4 61 protein alone and in conjunction with T4 41 protein. J. Biol. Chem. 1987, 262, 10873–10878. [Google Scholar] [PubMed]
- Cha, T.A.; Alberts, B.M. Effects of the bacteriophage T4 gene 41 and gene 32 proteins on rna primer synthesis: The coupling of leading-and lagging-strand DNA synthesis at a replication fork. Biochemistry 1990, 29, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xi, J.; Zhuang, Z.; Benkovic, S.J. The oligomeric T4 primase is the functional form during replication. J. Biol. Chem. 2005, 280, 25416–25423. [Google Scholar] [CrossRef] [PubMed]
- Huberman, J.A.; Kornberg, A.; Alberts, B.M. Stimulation of t4 bacteriophage DNA polymerase by the protein product of T4 gene 32. J. Mol. Biol. 1971, 62, 39–52. [Google Scholar] [CrossRef]
- Huang, C.; Hearst, J.; Alberts, B. Two types of replication proteins increase the rate at which T4 DNA polymerase traverses the helical regions in a single-stranded DNA template. J. Biol. Chem. 1981, 256, 4087–4094. [Google Scholar] [PubMed]
- Huang, C.-C.; Hearst, J.E. Pauses at positions of secondary structure during in vitro replication of single-stranded fd Bacteriophage DNA by T4 DNA polymerase. Anal. Biochem. 1980, 103, 127–139. [Google Scholar] [CrossRef]
- Shamoo, Y.; Friedman, A.M.; Parsons, M.R.; Konigsberg, W.H.; Steitz, T.A. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature 1995, 376, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Dudas, K.C.; Kreuzer, K.N. Bacteriophage T4 helicase loader protein gp59 functions as gatekeeper in origin-dependent replication in vivo. J. Biol. Chem. 2005, 280, 21561–21569. [Google Scholar] [CrossRef] [PubMed]
- Ishmael, F.T.; Alley, S.C.; Benkovic, S.J. Assembly of the Bacteriophage T4 helicase architecture and stoichiometry of the gp41-gp59 complex. J. Biol. Chem. 2002, 277, 20555–20562. [Google Scholar] [CrossRef] [PubMed]
- Delagoutte, E.; von Hippel, P.H. Mechanistic studies of the t4 DNA (gp41) replication helicase: Functional interactions of the c-terminal tails of the helicase subunits with the T4 (gp59) helicase loader protein. J. Mol. Biol. 2005, 347, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Chastain, P.D.; Makhov, A.M.; Nossal, N.G.; Griffith, J. Architecture of the replication complex and DNA loops at the fork generated by the bacteriophage t4 proteins. J. Biol. Chem. 2003, 278, 21276–21285. [Google Scholar] [CrossRef] [PubMed]
- Nossal, N.G.; Makhov, A.M.; Chastain, P.D.; Jones, C.E.; Griffith, J.D. Architecture of the Bacteriophage T4 replication complex revealed with nanoscale biopointers. J. Biol. Chem. 2007, 282, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, D.; Yue, H.; Spiering, M.M.; Zhao, C.; Benkovic, S.J.; Huang, T.J. Dark-field illumination on zero-mode waveguide/microfluidic hybrid chip reveals T4 replisomal protein interactions. Nano Lett. 2014, 14, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Mace, D.C.; Alberts, B.M. T4 DNA polymerase: Rates and processivity on single-stranded DNA templates. J. Mol. Biol. 1984, 177, 295–311. [Google Scholar] [CrossRef]
- Kelch, B.A.; Makino, D.L.; O’Donnell, M.; Kuriyan, J. How a DNA polymerase clamp loader opens a sliding clamp. Science 2011, 334, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Berdis, A.J.; Benkovic, S.J. Role of adenosine 5′-triphosphate hydrolysis in the assembly of the bacteriophage T4 DNA replication holoenzyme complex. Biochemistry 1996, 35, 9253–9265. [Google Scholar] [CrossRef] [PubMed]
- Perumal, S.K.; Ren, W.; Lee, T.-H.; Benkovic, S.J. How a holoenzyme for DNA replication is formed. Proc. Natl. Acad. Sci. USA 2013, 110, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K. Bacteriophage T4; Wiley Online Library: Washington, DC, USA, 1983. [Google Scholar]
- Kaboord, B.F.; Benkovic, S.J. Accessory proteins function as matchmakers in the assembly of the T4 DNA polymerase holoenzyme. Curr. Biol. 1995, 5, 149–157. [Google Scholar] [CrossRef]
- Schrock, R.D.; Alberts, B. Processivity of the gene 41 DNA helicase at the bacteriophage T4 DNA replication fork. J. Biol. Chem. 1996, 271, 16678–16682. [Google Scholar] [PubMed]
- Yang, J.; Zhuang, Z.; Roccasecca, R.M.; Trakselis, M.A.; Benkovic, S.J. The dynamic processivity of the T4 DNA polymerase during replication. Proc. Natl. Acad. Sci. USA 2004, 101, 8289–8294. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Hübscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Maul, R.W.; Scouten Ponticelli, S.K.; Duzen, J.M.; Sutton, M.D. Differential binding of Escherichia coli DNA polymerases to the β-sliding clamp. Mol. Microbial. 2007, 65, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Takahashi, M.; Hamdan, S.M.; Lee, S.-J.; Richardson, C.C. Exchange of DNA polymerases at the replication fork of bacteriophage T7. Proc. Natl. Acad. Sci. USA 2007, 104, 5312–5317. [Google Scholar] [CrossRef] [PubMed]
- Hacker, K.J.; Alberts, B.M. The rapid dissociation of the T4 DNA polymerase holoenzyme when stopped by a DNA hairpin helix. A model for polymerase release following the termination of each okazaki fragment. J. Biol. Chem. 1994, 269, 24221–24228. [Google Scholar] [PubMed]
- Cha, T.-A.; Alberts, B.M. The bacteriophage t4 DNA replication fork. Only DNA helicase is required for leading strand DNA synthesis by the DNA polymerase holoenzyme. J. Biol. Chem. 1989, 264, 12220–12225. [Google Scholar] [PubMed]
- Dong, F.; Weitzel, S.E.; Von Hippel, P.H. A coupled complex of T4 DNA replication helicase (gp41) and polymerase (gp43) can perform rapid and processive DNA strand-displacement synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 14456–14461. [Google Scholar] [CrossRef] [PubMed]
- Delagoutte, E.; von Hippel, P.H. Molecular mechanisms of the functional coupling of the helicase (gp41) and polymerase (gp43) of bacteriophage T4 within the DNA replication fork. Biochemistry 2001, 40, 4459–4477. [Google Scholar] [CrossRef] [PubMed]
- Ishmael, F.T.; Trakselis, M.A.; Benkovic, S.J. Protein-protein interactions in the bacteriophage T4 replisome the leading strand holoenzyme is physically linked to the lagging strand holoenzyme and the primosome. J. Biol. Chem. 2003, 278, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Stano, N.M.; Jeong, Y.-J.; Donmez, I.; Tummalapalli, P.; Levin, M.K.; Patel, S.S. DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature 2005, 435, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Manosas, M.; Spiering, M.M.; Ding, F.; Croquette, V.; Benkovic, S.J. Collaborative coupling between polymerase and helicase for leading-strand synthesis. Nucl. Acids Res. 2012, 40, 6187–6198. [Google Scholar] [CrossRef] [PubMed]
- Lionnet, T.; Spiering, M.M.; Benkovic, S.J.; Bensimon, D.; Croquette, V. Real-time observation of bacteriophage T4 gp41 helicase reveals an unwinding mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 19790–19795. [Google Scholar] [CrossRef] [PubMed]
- Salinas, F.; Benkovic, S.J. Characterization of bacteriophage T4-coordinated leading-and lagging-strand synthesis on a minicircle substrate. Proc. Natl. Acad. Sci. USA 2000, 97, 7196–7201. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Trakselis, M.A.; Roccasecca, R.M.; Benkovic, S.J. The application of a minicircle substrate in the study of the coordinated T4 DNA replication. J. Biol. Chem. 2003, 278, 49828–49838. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Beechem, J.M.; Patton, W.F. The utility of a two-color fluorescence electrophoretic mobility shift assay procedure for the analysis of DNA replication complexes. Electrophoresis 2004, 25, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.H.; Dong, F.; Latham, G.J.; von Hippel, P.H. Interactions of bacteriophage t4-coded primase (gp61) with the t4 replication helicase (gp41) and DNA in primosome formation. J. Biol. Chem. 1999, 274, 27287–27298. [Google Scholar] [CrossRef] [PubMed]
- Manosas, M.; Spiering, M.M.; Zhuang, Z.; Benkovic, S.J.; Croquette, V. Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat. Chem. Biol. 2009, 5, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-B.; Hite, R.K.; Hamdan, S.M.; Xie, X.S.; Richardson, C.C.; Van Oijen, A.M. DNA T4 primase acts as a molecular brake in DNA replication. Nature 2006, 439, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Syed, S.; Donmez, I.; Patel, G.; Ha, T.; Patel, S.S. Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature 2009, 462, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Drake, J.W. Conditional coupling of leading-strand and lagging-strand DNA synthesis at bacteriophage T4 replication forks. J. Biol. Chem. 2001, 276, 29559–29566. [Google Scholar] [CrossRef] [PubMed]
- Trakselis, M.A.; Roccasecca, R.M.; Yang, J.; Valentine, A.M.; Benkovic, S.J. Dissociative properties of the proteins within the bacteriophage T4 replisome. J. Biol. Chem. 2003, 278, 49839–49849. [Google Scholar] [CrossRef] [PubMed]
- Trakselis, M.A.; Alley, S.C.; Abel-Santos, E.; Benkovic, S.J. Creating a dynamic picture of the sliding clamp during T4 DNA polymerase holoenzyme assembly by using fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2001, 98, 8368–8375. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.W.; Kumar, R.; Benkovic, S.J. Rna primer handoff in bacteriophage T4 DNA replication the role of single-stranded DNA-binding protein and polymerase accessory proteins. J. Biol. Chem. 2008, 283, 22838–22846. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nelson, S.W.; Benkovic, S.J. The control mechanism for lagging strand polymerase recycling during bacteriophage T4 DNA replication. Mol. Cell 2006, 21, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.E.; Sexton, D.J.; Benkovic, S.J. Dissociation of bacteriophage t4 DNA polymerase and its processivity clamp after completion of okazaki fragment synthesis. Biochemistry 1997, 36, 14409–14417. [Google Scholar] [CrossRef] [PubMed]
- Tougu, K.; Marians, K.J. The interaction between helicase and primase sets the replication fork clock. J. Biol. Chem. 1996, 271, 21398–21405. [Google Scholar] [PubMed]
- Kurth, I.; Georgescu, R.E.; O’Donnell, M.E. A solution to release twisted DNA during chromosome replication by coupled DNA polymerases. Nature 2013, 496, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; McHenry, C.S. Cycling of the E. coli lagging strand polymerase is triggered exclusively by the availability of a new primer at the replication fork. Nucl. Acids Res. 2014, 42, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Geertsema, H.J.; van Oijen, A.M. A single-molecule view of DNA replication: The dynamic nature of multi-protein complexes revealed. Curr. Opin. Struct. Biol. 2013, 23, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yue, H.; Spiering, M.M.; Benkovic, S.J. Insights into okazaki fragment synthesis by the T4 replisome the fate of lagging-strand holoenzyme components and their influence on Okazaki fragment size. J. Biol. Chem. 2013, 288, 20807–20816. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noble, E.; Spiering, M.M.; Benkovic, S.J. Coordinated DNA Replication by the Bacteriophage T4 Replisome. Viruses 2015, 7, 3186-3200. https://doi.org/10.3390/v7062766
Noble E, Spiering MM, Benkovic SJ. Coordinated DNA Replication by the Bacteriophage T4 Replisome. Viruses. 2015; 7(6):3186-3200. https://doi.org/10.3390/v7062766
Chicago/Turabian StyleNoble, Erin, Michelle M. Spiering, and Stephen J. Benkovic. 2015. "Coordinated DNA Replication by the Bacteriophage T4 Replisome" Viruses 7, no. 6: 3186-3200. https://doi.org/10.3390/v7062766
APA StyleNoble, E., Spiering, M. M., & Benkovic, S. J. (2015). Coordinated DNA Replication by the Bacteriophage T4 Replisome. Viruses, 7(6), 3186-3200. https://doi.org/10.3390/v7062766