
High-Risk Human Papillomavirus Targets Crossroads in Immune Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Viral Recognition by Keratinocytes
3. HPV Influences Innate Immune Signaling
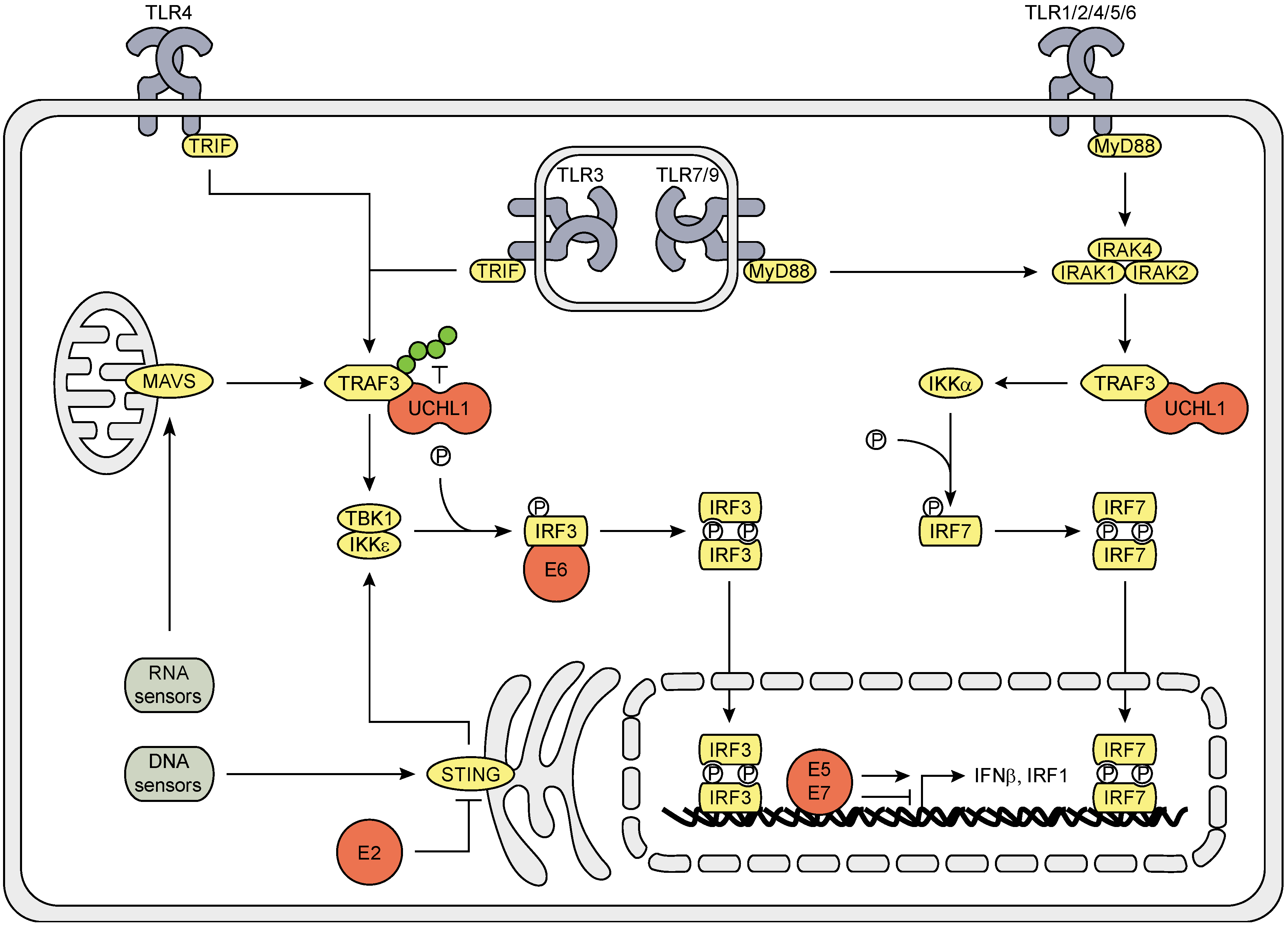
3.1. The Effect of HPV (Proteins) on the IRF Signaling Pathway
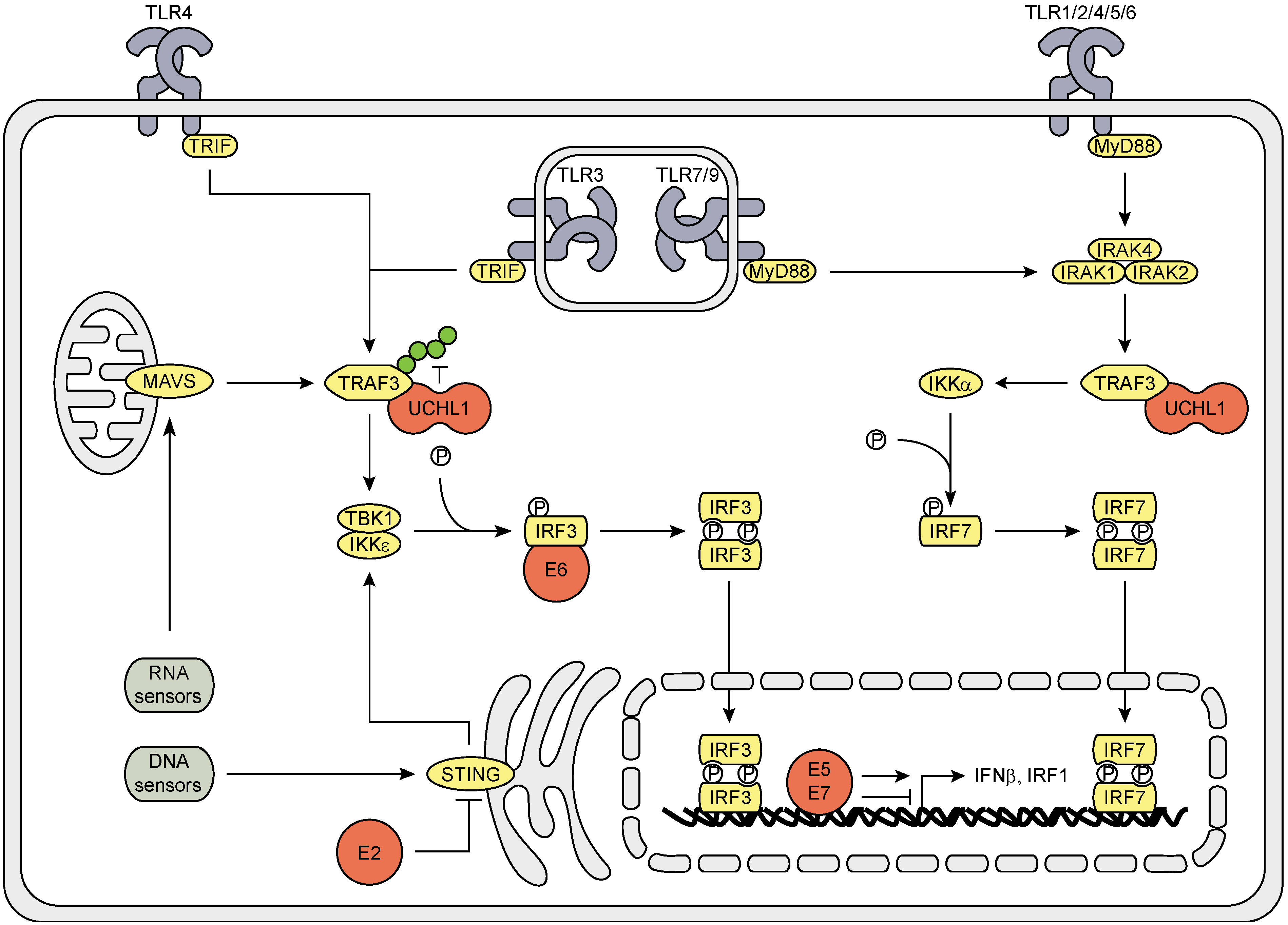
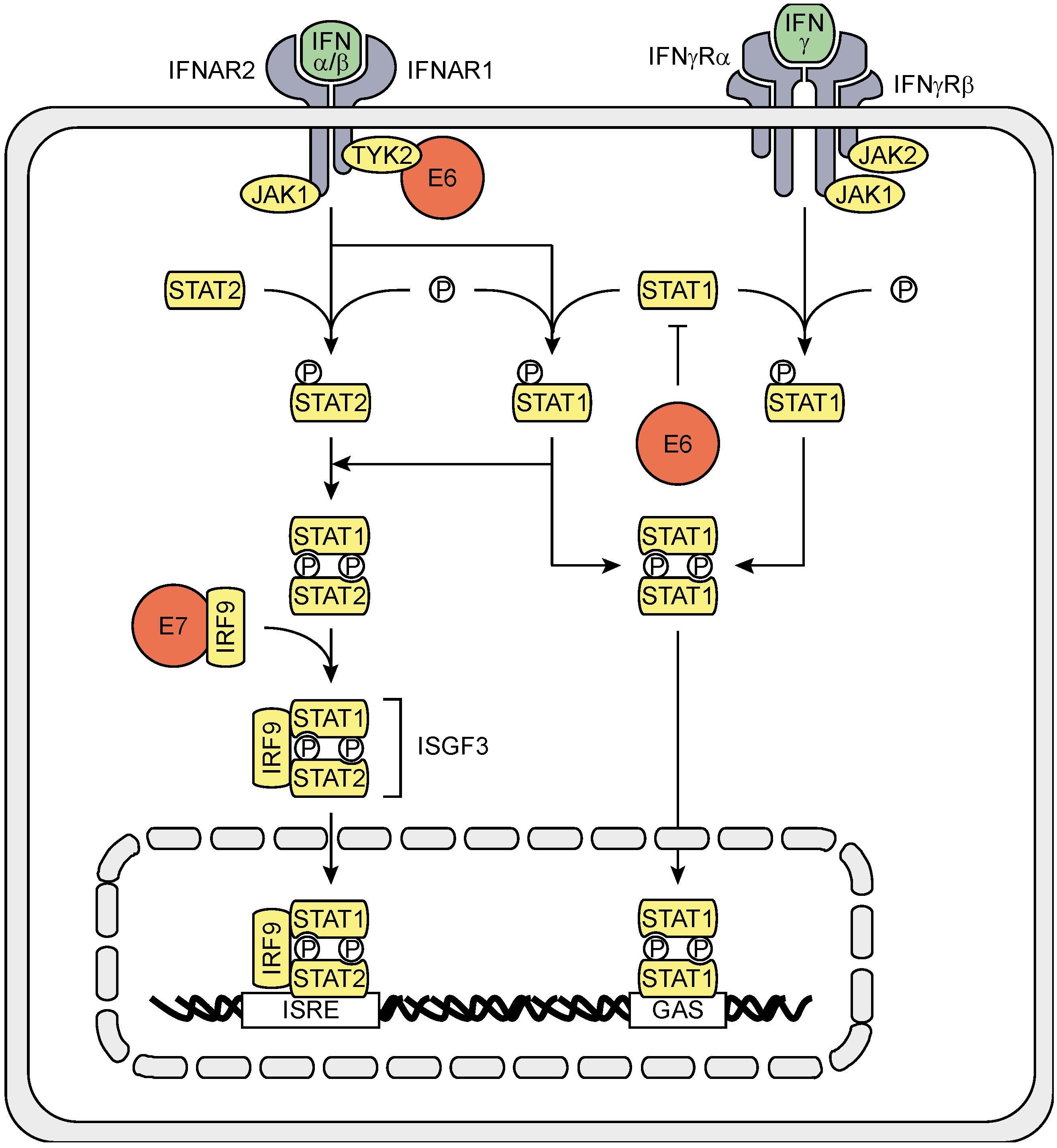
3.2. The Effects of HPV (Proteins) on IFNAR Signaling

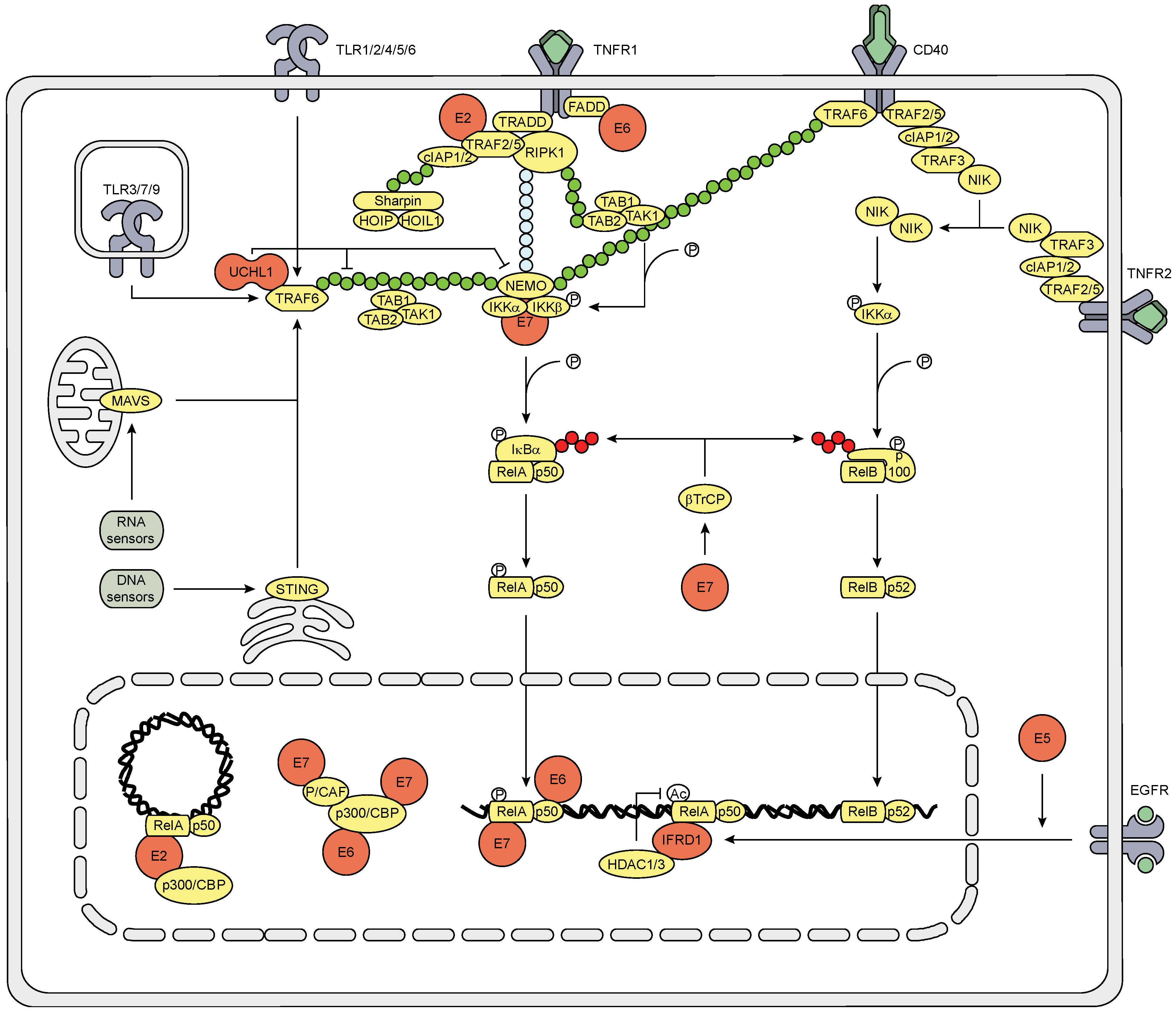
3.3. The Effect of HPV (Proteins) on the NFκB Signaling Pathway
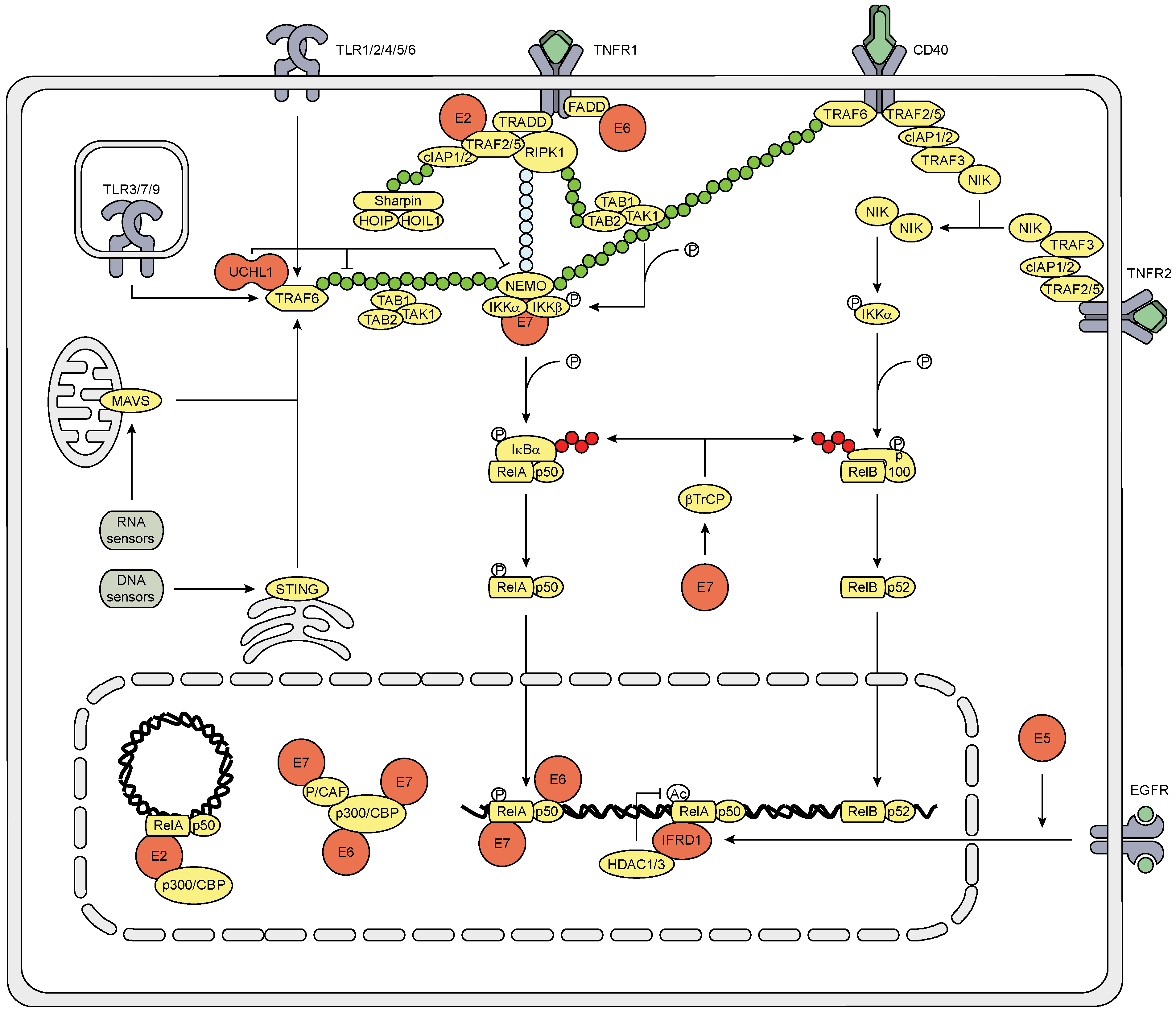
3.4. The Effect on the Inflammasome Pathway
4. The Action of KCs to Secondary Immune Signals is Suppressed by HPV
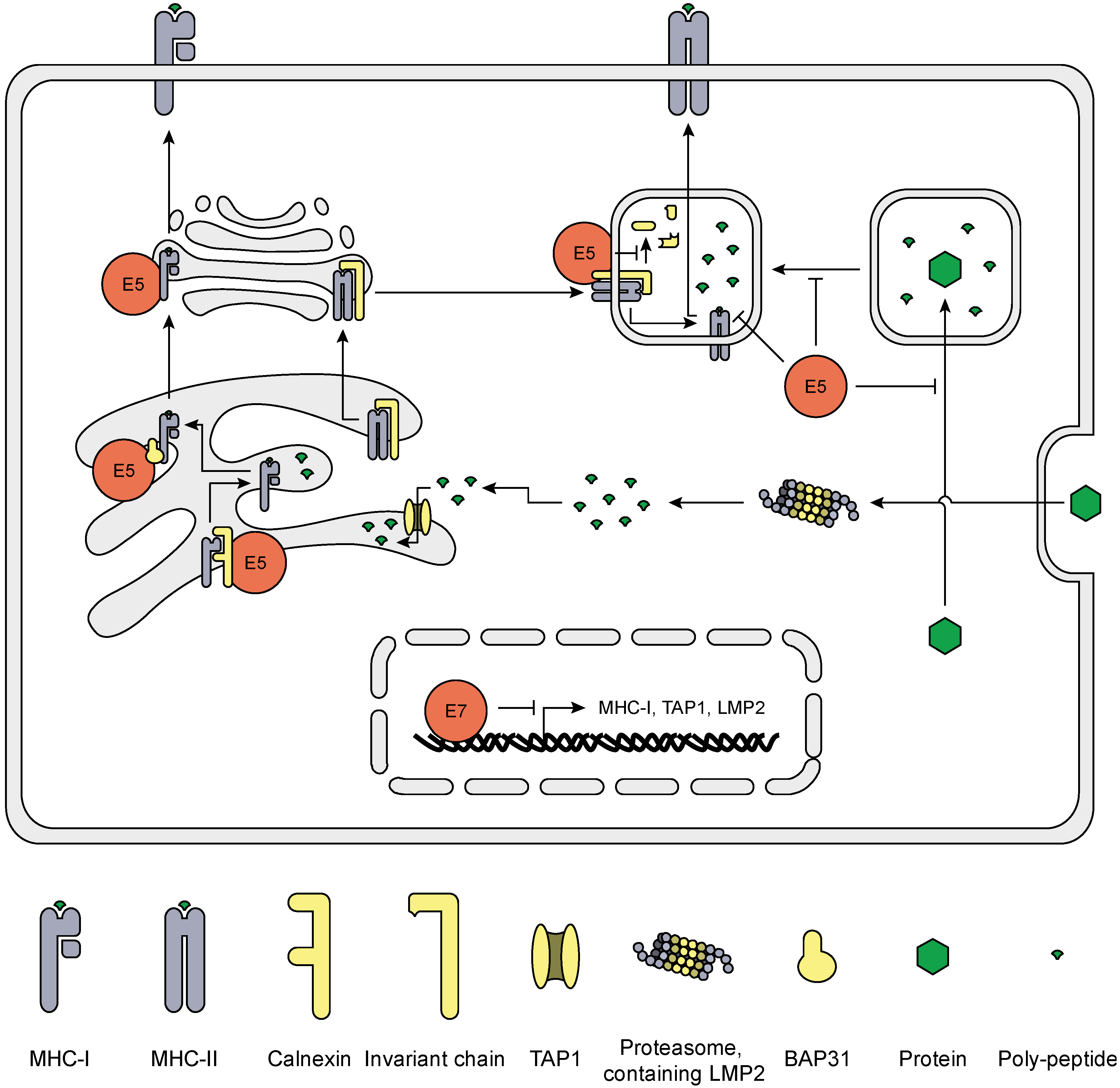
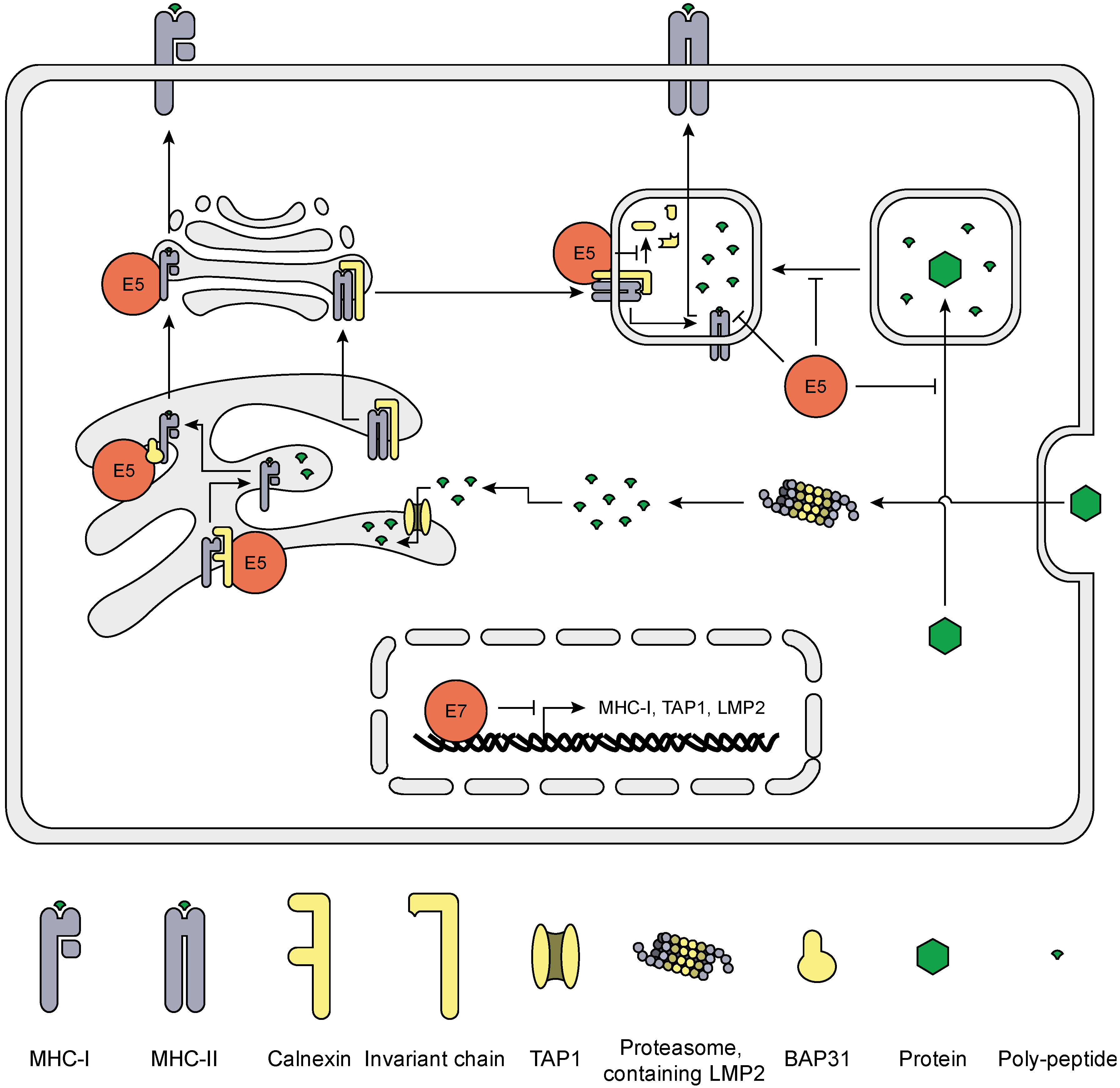
5. HrHPV Influences MHC Surface Expression and Peptide Presentation
6. Final Comments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Cubie, H.A. Diseases associated with human papillomavirus infection. Virology 2013, 445, 21–34. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Akgul, B.; Cooke, J.C.; Storey, A. HPV-associated skin disease. J. Pathol. 2006, 208, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Veldhuijzen, N.J.; Snijders, P.J.; Reiss, P.; Meijer, C.J.; van de Wijgert, J.H. Factors affecting transmission of mucosal human papillomavirus. Lancet Infect. Dis. 2010, 10, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Anttila, A.; Jordan, J.; Ronco, G.; Schenck, U.; Segnan, N.; Wiener, H.; Herbert, A.; von Karsa, L. European Guidelines for Quality Assurance in Cervical Cancer Screening. Second edition—Summary document. Ann. Oncol. 2010, 21, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 2007, 121, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Bienkowska-Haba, M. Viral entry mechanisms: Human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J. 2009, 276, 7206–7216. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32 (Suppl. 1), S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Bedell, M.A.; Hudson, J.B.; Golub, T.R.; Turyk, M.E.; Hosken, M.; Wilbanks, G.D.; Laimins, L.A. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 1991, 65, 2254–2260. [Google Scholar] [PubMed]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Wang, S. E3 ubiquitin ligases and human papillomavirus-induced carcinogenesis. J. Int. Med. Res. 2014, 42, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Melief, C.J. Therapeutic vaccination against human papilloma virus induced malignancies. Curr. Opin. Immunol. 2011, 23, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [PubMed]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLOS Pathog. 2013, 9, e1003384. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLOS ONE 2011, 6, e17848. [Google Scholar] [CrossRef] [PubMed]
- Reinholz, M.; Kawakami, Y.; Salzer, S.; Kreuter, A.; Dombrowski, Y.; Koglin, S.; Kresse, S.; Ruzicka, T.; Schauber, J. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch. Dermatol. Res. 2013, 305, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Kalali, B.N.; Kollisch, G.; Mages, J.; Muller, T.; Bauer, S.; Wagner, H.; Ring, J.; Lang, R.; Mempel, M.; Ollert, M. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J. Immunol. 2008, 181, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Munch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Muller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.M.; Al-Khairy, D.; Ingalls, R.R. Innate immunity at the mucosal surface: Role of toll-like receptor 3 and toll-like receptor 9 in cervical epithelial cell responses to microbial pathogens. Biol. Reprod. 2006, 74, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.E.; Laimins, L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef] [PubMed]
- Karstensen, B.; Poppelreuther, S.; Bonin, M.; Walter, M.; Iftner, T.; Stubenrauch, F. Gene expression profiles reveal an upregulation of E2F and downregulation of interferon targets by HPV18 but no changes between keratinocytes with integrated or episomal viral genomes. Virology 2006, 353, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-kappa transcription in keratinocytes. PLOS One 2014, 9, e91473. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [PubMed]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Massimi, P.; Banks, L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int. J. Mol. Med. 2000, 5, 661–666. [Google Scholar] [PubMed]
- Muto, V.; Stellacci, E.; Lamberti, A.G.; Perrotti, E.; Carrabba, A.; Matera, G.; Sgarbanti, M.; Battistini, A.; Liberto, M.C.; Foca, A. Human papillomavirus type 16 E5 protein induces expression of beta interferon through interferon regulatory factor 1 in human keratinocytes. J. Virol. 2011, 85, 5070–5080. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.; Pellegrini, S.; Matlashewski, G.J.; Koromilas, A.E. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Barnard, P.; McMillan, N.A. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology 1999, 259, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Barnard, P.; Payne, E.; McMillan, N.A. The human papillomavirus E7 protein is able to inhibit the antiviral and anti-growth functions of interferon-alpha. Virology 2000, 277, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Havard, L.; Delvenne, P.; Frare, P.; Boniver, J.; Giannini, S.L. Differential production of cytokines and activation of NF-kappaB in HPV-transformed keratinocytes. Virology 2002, 298, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Havard, L.; Rahmouni, S.; Boniver, J.; Delvenne, P. High levels of p105 (NFKB1) and p100 (NFKB2) proteins in HPV16-transformed keratinocytes: Role of E6 and E7 oncoproteins. Virology 2005, 331, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Prabhavathy, D.; Prabhakar, B.N.; Karunagaran, D. HPV16 E2-mediated potentiation of NF-kappaB activation induced by TNF-alpha involves parallel activation of STAT3 with a reduction in E2-induced apoptosis. Mol. Cell. Biochem. 2014, 394, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Vancurova, I.; Wu, R.; Miskolci, V.; Sun, S. Increased p50/p50 NF-kappaB activation in human papillomavirus type 6- or type 11-induced laryngeal papilloma tissue. J. Virol. 2002, 76, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Takami, Y.; Nakagami, H.; Morishita, R.; Katsuya, T.; Cui, T.X.; Ichikawa, T.; Saito, Y.; Hayashi, H.; Kikuchi, Y.; Nishikawa, T.; et al. Ubiquitin carboxyl-terminal hydrolase L1, a novel deubiquitinating enzyme in the vasculature, attenuates NF-kappaB activation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Spitkovsky, D.; Hehner, S.P.; Hofmann, T.G.; Moller, A.; Schmitz, M.L. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa B kinase complex. J. Biol. Chem. 2002, 277, 25576–25582. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- Huang, S.M.; McCance, D.J. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J. Virol. 2002, 76, 8710–8721. [Google Scholar] [CrossRef] [PubMed]
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 2003, 22, 7871–7881. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, B.; Kim, J.; Kim, D.W.; Choe, J. cAMP response element-binding protein-binding protein binds to human papillomavirus E2 protein and activates E2-dependent transcription. J. Biol. Chem. 2000, 275, 7045–7051. [Google Scholar] [CrossRef] [PubMed]
- Marcello, A.; Massimi, P.; Banks, L.; Giacca, M. Adeno-associated virus type 2 rep protein inhibits human papillomavirus type 16 E2 recruitment of the transcriptional coactivator p300. J. Virol. 2000, 74, 9090–9098. [Google Scholar] [CrossRef] [PubMed]
- Akerman, G.S.; Tolleson, W.H.; Brown, K.L.; Zyzak, L.L.; Mourateva, E.; Engin, T.S.; Basaraba, A.; Coker, A.L.; Creek, K.E.; Pirisi, L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001, 61, 3837–3843. [Google Scholar] [PubMed]
- Tummers, B.; Goedemans, R.; Pelascini, L.P.L.; Jordanova, E.S.; van Esch, E.M.G.; Meyers, C.; Melief, C.J.M.; Boer, J.M.; van der Burg, S.H. The interferon-related developmental regulator 1 is used by human papillomavirus to suppress NFκB activation. Nat. Commun. 2015, 6, 6537. [Google Scholar] [CrossRef]
- Kim, S.H.; Juhnn, Y.S.; Kang, S.; Park, S.W.; Sung, M.W.; Bang, Y.J.; Song, Y.S. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ ERK1,2 and PI3K/Akt. Cell. Mol. Life Sci. 2006, 63, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The HPV16 E6 oncoprotein causes prolonged receptor protein tyrosine kinase signaling and enhances internalization of phosphorylated receptor species. PLOS Pathog. 2013, 9, e1003237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, A.; Ueki, I.; Min-Oo, G.; Ballon-Landa, E.; Knoff, D.; Galen, B.; Lanier, L.L.; Nadel, J.A.; Koff, J.L. EGFR activation suppresses respiratory virus-induced IRF1-dependent CXCL10 production. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L186–L196. [Google Scholar] [CrossRef] [PubMed]
- Mascia, F.; Mariani, V.; Girolomoni, G.; Pastore, S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am. J. Pathol. 2003, 163, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Paul, T.; Schumann, C.; Rudiger, S.; Boeck, S.; Heinemann, V.; Kachele, V.; Steffens, M.; Scholl, C.; Hichert, V.; Seufferlein, T.; et al. Cytokine regulation by epidermal growth factor receptor inhibitors and epidermal growth factor receptor inhibitor associated skin toxicity in cancer patients. Eur. J. Cancer 2014, 50, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Niebler, M.; Qian, X.; Hofler, D.; Kogosov, V.; Kaewprag, J.; Kaufmann, A.M.; Ly, R.; Bohmer, G.; Zawatzky, R.; Rosl, F.; et al. Post-translational control of IL-1beta via the human papillomavirus type 16 E6 oncoprotein: A novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLOS Pathog. 2013, 9, e1003536. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Melief, C.J. Immunotherapy for persistent viral infections and associated disease. Trends Immunol. 2011, 32, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef] [PubMed]
- Boulabiar, M.; Boubaker, S.; Favre, M.; Demeret, C. Keratinocyte sensitization to tumour necrosis factor-induced nuclear factor kappa B activation by the E2 regulatory protein of human papillomaviruses. J. Gen. Virol. 2011, 92, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; van Esch, E.M.; Ma, W.; Goedemans, R.; Pelascini, L.P.L.; Vilarrasa-Blasi, R.; Torreggiani, S.; Meyers, C.; Melief, C.J.M.; Boer, J.M.; et al. Human papillomavirus (HPV) down regulates the expression of IFITM1 to resist the anti-proliferative effects of IFNγ and TNFα. 2015; submitted. [Google Scholar]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Matsumoto, A.; Inuzuka, H.; Zhai, B.; Lau, A.W.; Wan, L.; Gao, D.; Shaik, S.; Yuan, M.; Gygi, S.P.; et al. SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep. 2012, 1, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Galy, A.H.; Spits, H. CD40 is functionally expressed on human thymic epithelial cells. J. Immunol. 1992, 149, 775–782. [Google Scholar] [PubMed]
- Tummers, B.; Goedemans, R.; Jha, V.; Meyers, C.; Melief, C.J.M.; van der Burg, S.H.; Boer, J.M. CD40-mediated amplification of local immunity by epithelial cells is impaired by HPV. J. Investig. Dermatol. 2014, 134, 2918–2927. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Frazer, I.H.; Leggatt, G.R. Keratinocytes efficiently process endogenous antigens for cytotoxic T-cell mediated lysis. Exp. Dermatol. 2009, 18, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S.; Graham, S.V.; Cortese, M.S.; Ashrafi, G.H.; Araibi, E.H.; Dornan, E.S.; Miners, K.; Nunes, C.; Man, S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010, 407, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Leggatt, G.R.; Frazer, I.H. Human papillomavirus 16 E7 protein inhibits interferon-gamma-mediated enhancement of keratinocyte antigen processing and T-cell lysis. FEBS J. 2011, 278, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene 2000, 19, 4930–4935. [Google Scholar] [CrossRef] [PubMed]
- Heller, C.; Weisser, T.; Mueller-Schickert, A.; Rufer, E.; Hoh, A.; Leonhardt, R.M.; Knittler, M.R. Identification of key amino acid residues that determine the ability of high risk HPV16-E7 to dysregulate major histocompatibility complex class I expression. J. Biol. Chem. 2011, 286, 10983–10997. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ou, X.; Xiong, J.; Wang, T. HPV16E7 mediates HADC chromatin repression and downregulation of MHC class I genes in HPV16 tumorigenic cells through interaction with an MHC class I promoter. Biochem. Biophys. Res. Commun. 2006, 349, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhan, T.; Li, C.; Liu, M.; Wang, Q.K. Repression of MHC class I transcription by HPV16E7 through interaction with a putative RXRbeta motif and NF-kappaB cytoplasmic sequestration. Biochem. Biophys. Res. Commun. 2009, 388, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, J.; Zhao, K.N. Human papillomavirus 16-encoded E7 protein inhibits IFN-gamma-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J. Gen. Virol. 2013, 94, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Ashrafi, G.H.; Campo, M.S. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 2010, 126, 1675–1682. [Google Scholar] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Gruener, M.; Bravo, I.G.; Momburg, F.; Alonso, A.; Tomakidi, P. The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol. J. 2007, 4, e116. [Google Scholar] [CrossRef]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.A.; Laimins, L.A. Bap31 is a novel target of the human papillomavirus E5 protein. J. Virol. 2008, 82, 10042–10051. [Google Scholar] [CrossRef] [PubMed]
- Abe, F.; van Prooyen, N.; Ladasky, J.J.; Edidin, M. Interaction of Bap31 and MHC class I molecules and their traffic out of the endoplasmic reticulum. J. Immunol. 2009, 182, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.W.; Blum, J.S.; Roman, A. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [PubMed]
- Thomsen, P.; van Deurs, B.; Norrild, B.; Kayser, L. The HPV16 E5 oncogene inhibits endocytic trafficking. Oncogene 2000, 19, 6023–6032. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.E.; Pena, L.; Sen, G.C.; Park, J.K.; Laimins, L.A. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J. Virol. 2002, 76, 8864–8874. [Google Scholar] [CrossRef] [PubMed]
- Herdman, M.T.; Pett, M.R.; Roberts, I.; Alazawi, W.O.; Teschendorff, A.E.; Zhang, X.Y.; Stanley, M.A.; Coleman, N. Interferon-beta treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Saikia, P.; Sen, G.C. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 2008, 27, 3311–3321. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fontela, C.; Pazos, M.; Delgado, I.; Murk, W.; Mungamuri, S.K.; Lee, S.W.; Garcia-Sastre, A.; Moran, T.M.; Aaronson, S.A. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J. Immunol. 2011, 187, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Branca, M.; Giorgi, C.; Ciotti, M.; Santini, D.; di Bonito, L.; Costa, S.; Benedetto, A.; Bonifacio, D.; Di Bonito, P.; Paba, P.; et al. Upregulation of nuclear factor-kappaB (NF-kappaB) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus or disease outcome in cervical cancer. Diagn. Cytopathol. 2006, 34, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Katzenellenbogen, R.A.; Grandori, C.; Galloway, D.A. NFX1 plays a role in human papillomavirus type 16 E6 activation of NFkappaB activity. J. Virol. 2010, 84, 11461–11469. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Mo, D.; Liu, H.; Veena, M.S.; Srivatsan, E.S.; Massoumi, R.; Rettig, M.B. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 2008, 14, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Vandermark, E.R.; Deluca, K.A.; Gardner, C.R.; Marker, D.F.; Schreiner, C.N.; Strickland, D.A.; Wilton, K.M.; Mondal, S.; Woodworth, C.D. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 2012, 425, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, P.K.; Lichtenstein, P.; Gyllensten, U.B. Heritability of cervical tumours. Int. J. Cancer 2000, 88, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.M.; Jordanova, E.S.; van Wezel, T.; Uh, H.W.; Corver, W.E.; Kwappenberg, K.M.; Verduijn, W.; Kenter, G.G.; van der Burg, S.H.; Fleuren, G.J. Genetic variation of antigen processing machinery components and association with cervical carcinoma. Genes Chromosom. Cancer 2007, 46, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Bratti, M.C.; Rodriguez, A.C.; Herrero, R.; Burk, R.D.; Porras, C.; Gonzalez, P.; Sherman, M.E.; Wacholder, S.; Lan, Z.E.; et al. Common variants in immune and DNA repair genes and risk for human papillomavirus persistence and progression to cervical cancer. J. Infect. Dis. 2009, 199, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Madeleine, M.M.; Johnson, L.G.; Du, Q.; Galloway, D.A.; Malkki, M.; Petersdorf, E.W.; Schwartz, S.M. Genetic variation in the TLR and NF-kappaB pathways and cervical and vulvar cancer risk: A population-based case-control study. Int. J. Cancer 2014, 134, 437–444. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tummers, B.; Van der Burg, S.H. High-Risk Human Papillomavirus Targets Crossroads in Immune Signaling. Viruses 2015, 7, 2485-2506. https://doi.org/10.3390/v7052485
Tummers B, Van der Burg SH. High-Risk Human Papillomavirus Targets Crossroads in Immune Signaling. Viruses. 2015; 7(5):2485-2506. https://doi.org/10.3390/v7052485
Chicago/Turabian StyleTummers, Bart, and Sjoerd H. Van der Burg. 2015. "High-Risk Human Papillomavirus Targets Crossroads in Immune Signaling" Viruses 7, no. 5: 2485-2506. https://doi.org/10.3390/v7052485
APA StyleTummers, B., & Van der Burg, S. H. (2015). High-Risk Human Papillomavirus Targets Crossroads in Immune Signaling. Viruses, 7(5), 2485-2506. https://doi.org/10.3390/v7052485