
The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
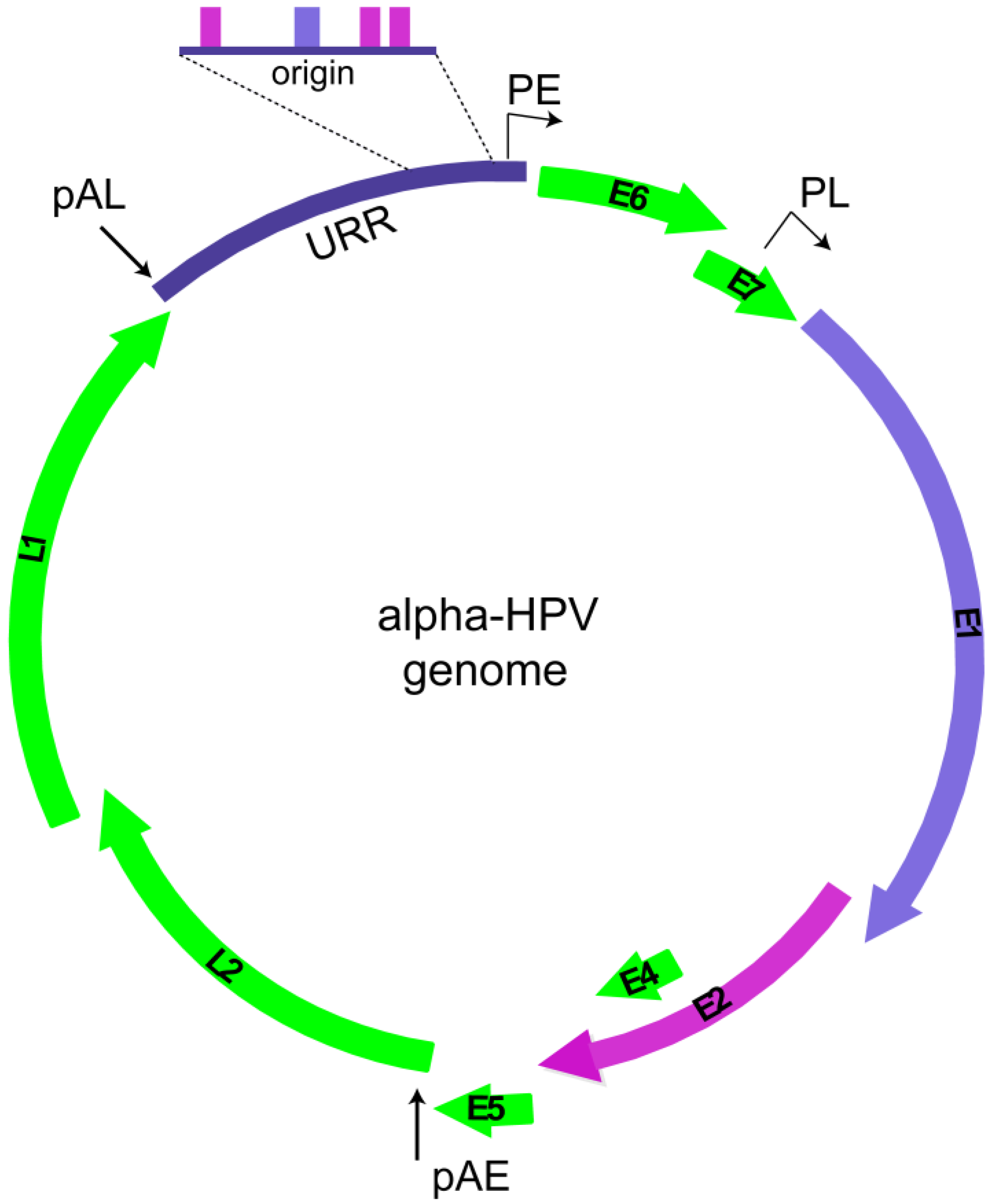
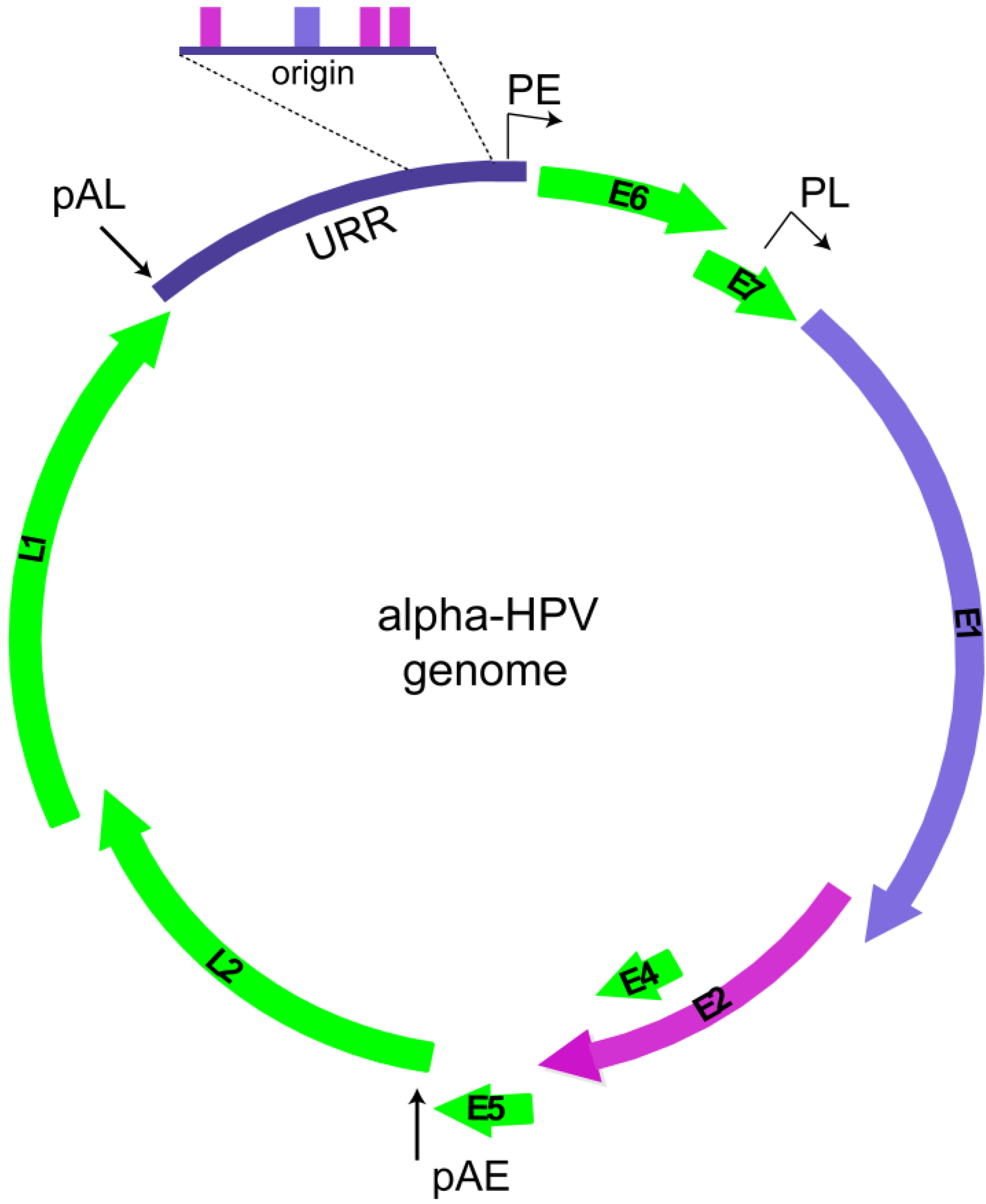
2. Replication Phases in the Papillomavirus Life Cycle
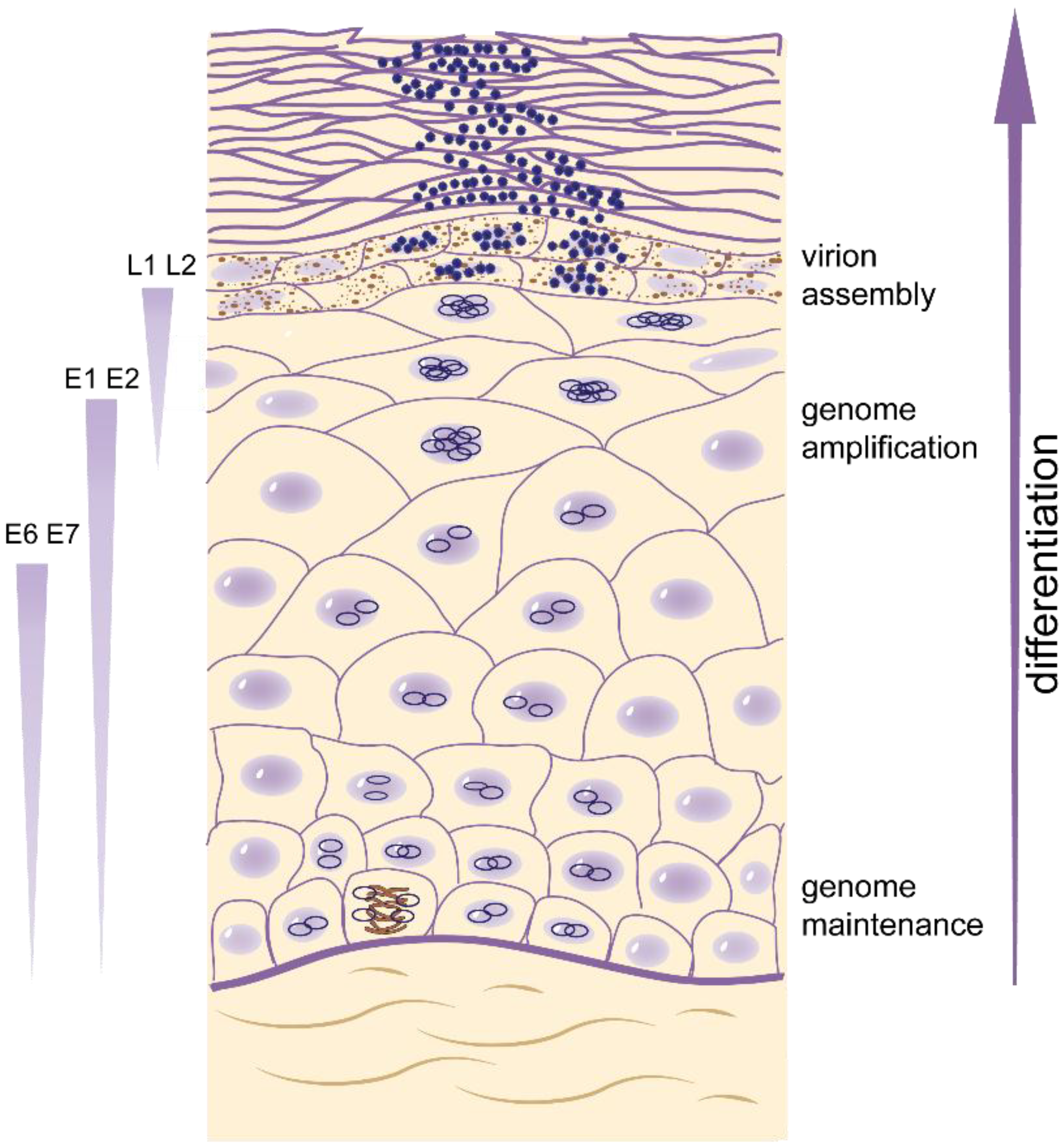
2.1. Overview of Phases of Replication in Papillomavirus Infection

2.2. HPV Replication Proteins
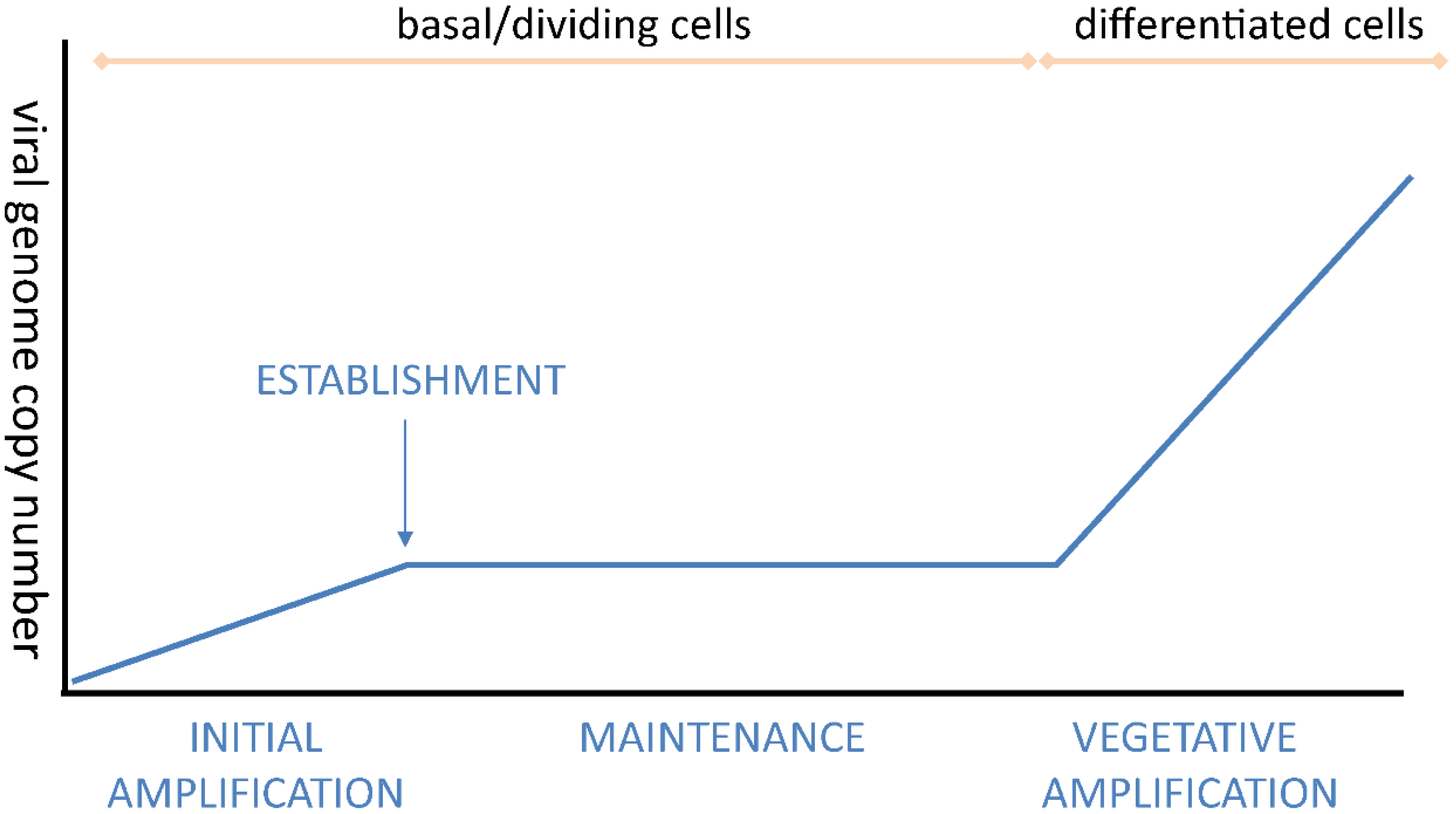
2.3. Initial Amplification and Establishment
2.4. Maintenance Replication and Persistence

2.5. Differentiation-Dependent Viral DNA Amplification
3. Role of the DNA Damage Response (DDR) in HPV Replication
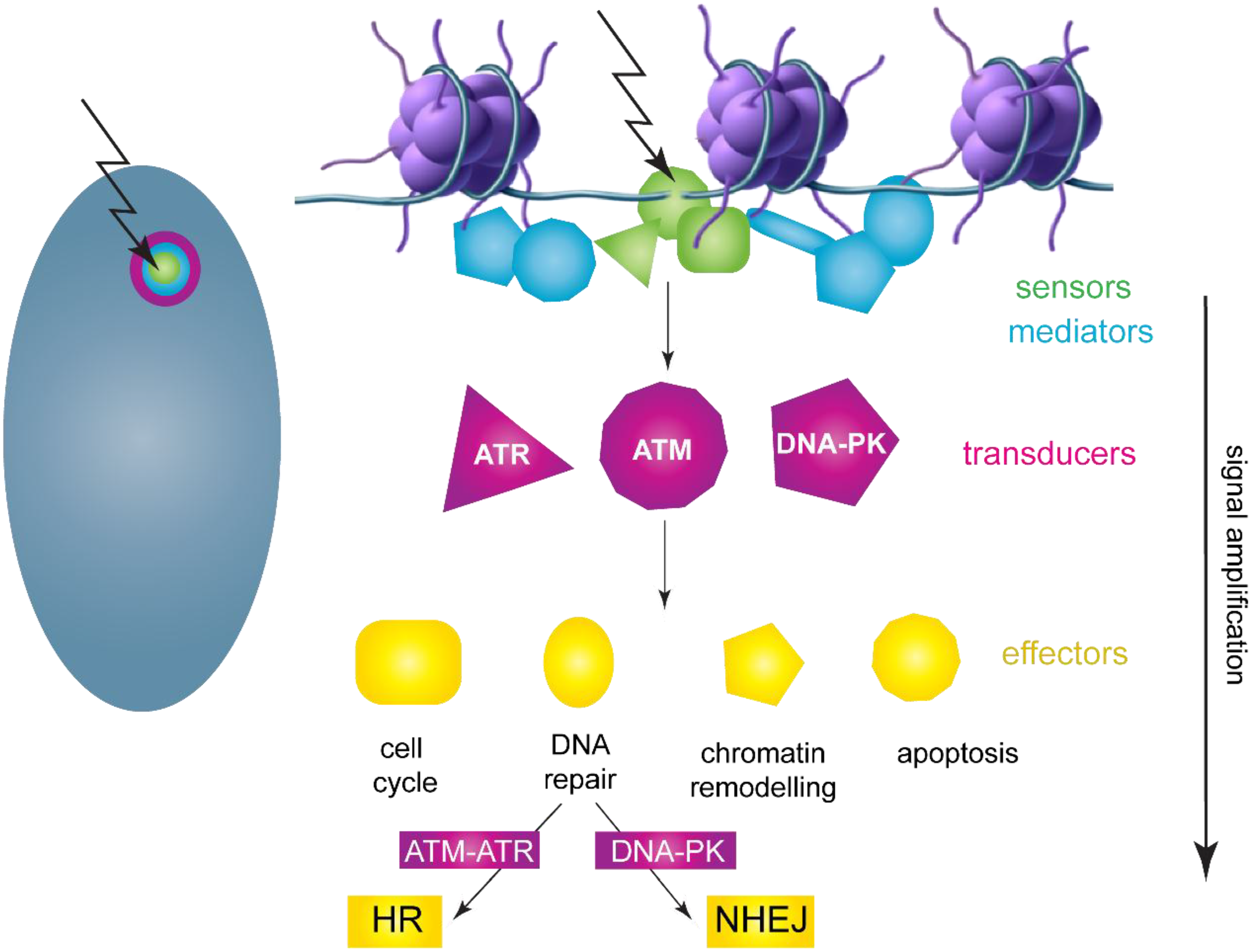
3.1. DNA Damage Response Overview
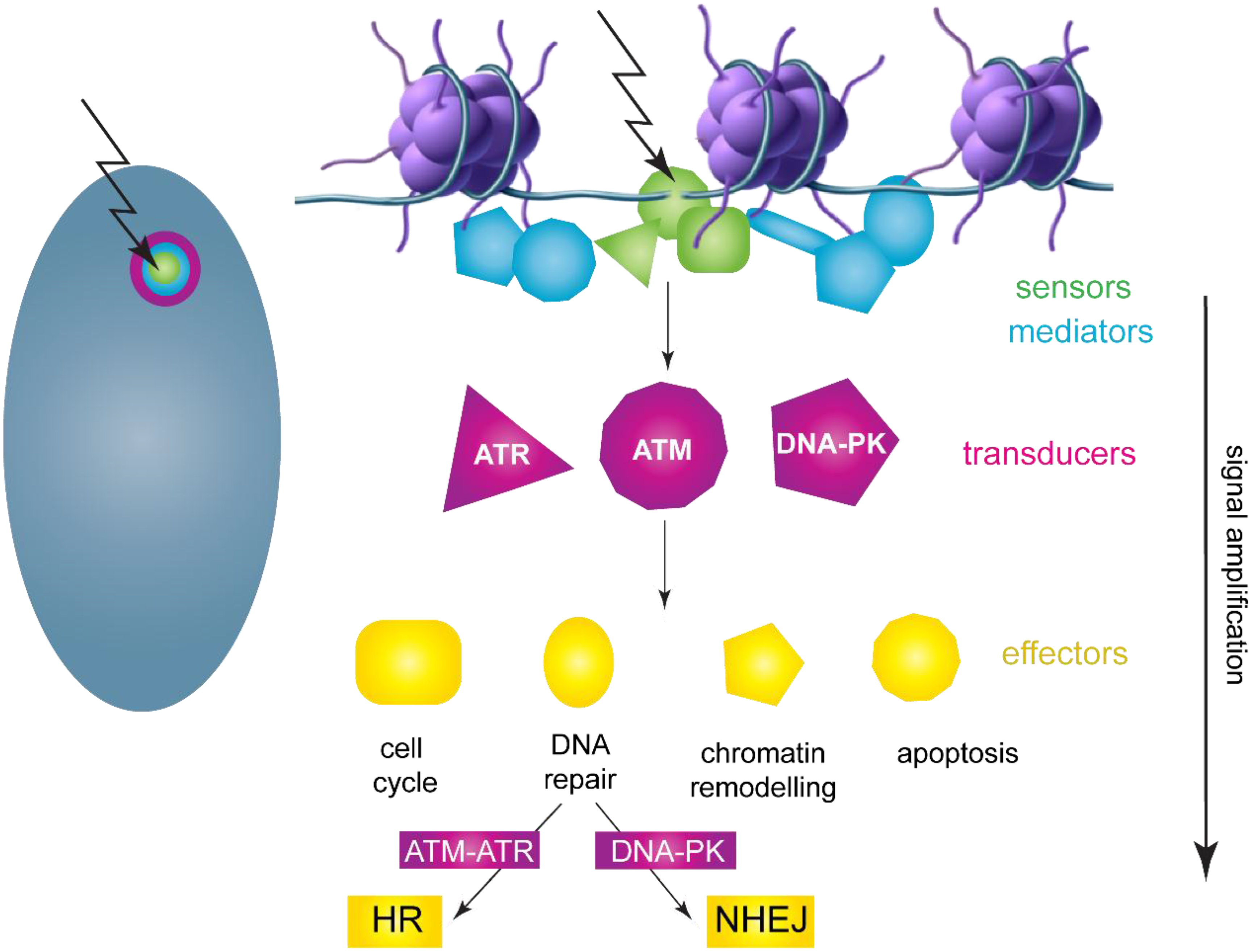
3.2. HPV Replication Foci Formation
3.3. The Role of the DNA Damage Response during Initial Amplification of HPV
3.4. The Role of the DNA Damage Response during Maintenance Replication.
3.5. The Role of the DNA Damage Response during Vegetative Amplification of Viral Genomes
3.6. The Role of the DNA Damage Response in HPV Integration and Carcinogenesis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cubie, H.A. Diseases associated with human papillomavirus infection. Virology 2013, 445, 21–34. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomavirus infections—A major cause of human cancers. Biochim. Biophys. Acta 1996, 1288, F55–F78. [Google Scholar] [PubMed]
- HPV-Associated Cancers Statistics. Available online: http://www.cdc.gov/cancer/hpv/statistics/ (accessed on 13 March 2015).
- NIAID The Papillomavirus Episteme. Available online: http://pave.niaid.nih.gov/ (accessed on 13 March 2015).
- McBride, A.A. Replication and partitioning of papillomavirus genomes. Adv. Virus Res. 2008, 72, 155–205. [Google Scholar] [PubMed]
- Mohr, I.J.; Clark, R.; Sun, S.; Androphy, E.J.; MacPherson, P.; Botchan, M.R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 1990, 250, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Ustav, M.; Stenlund, A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 1991, 10, 449–457. [Google Scholar] [PubMed]
- Ustav, M.; Ustav, E.; Szymanski, P.; Stenlund, A. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J. 1991, 10, 4321–4329. [Google Scholar] [PubMed]
- Sanders, C.M.; Stenlund, A. Recruitment and loading of the E1 initiator protein: An ATP-dependent process catalysed by a transcription factor. EMBO J. 1998, 17, 7044–7055. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Nakahara, T.; Ohno, S.; Narisawa-Saito, M.; Yugawa, T.; Fujita, M.; Yamato, K.; Natori, Y.; Kiyono, T. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol. 2012, 86, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lambert, P.F. E1 protein of bovine papillomavirus 1 is not required for the maintenance of viral plasmid DNA replication. Virology 2002, 293, 10–14. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Bromberg-White, J.L.; Meyers, C. The role of the human papillomavirus type 18 E7 oncoprotein during the complete viral life cycle. Virology 2005, 338, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Duffy, A.A.; Wang, H.K.; Broker, T.R. A highly efficient system to produce infectious human papillomavirus: Elucidation of natural virus-host interactions. Cell Cycle 2009, 8, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Wang, H.K.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 2011, 286, 15473–15482. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human papillomavirus type 16 E1^E4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 2005, 79, 13150–13165. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Schelhaas, M. Concepts of papillomavirus entry into host cells. Curr. Opin. Virol. 2014, 4, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Budgeon, L.R.; Christensen, N.D. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 2006, 347, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.F.; Aliesky, H.A.; Cooper, K. Optimization of biotinyl-tyramide-based in situ hybridization for sensitive background-free applications on formalin-fixed, paraffin-embedded tissue specimens. BMC Clin. Pathol. 2003, 3, e2. [Google Scholar] [CrossRef]
- Peh, W.L.; Middleton, K.; Christensen, N.; Nicholls, P.; Egawa, K.; Sotlar, K.; Brandsma, J.; Percival, A.; Lewis, J.; Liu, W.J.; et al. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 2002, 76, 10401–10416. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Hummel, M.; Iftner, T.; Laimins, L.A. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 2000, 74, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Botchan, M.; Berg, L.; Reynolds, J.; Lusky, M. The bovine papillomavirus replicon. In Papillomaviruses; John Wiley & Sons: New York, NY, USA, 1986; pp. 53–67. [Google Scholar]
- Roberts, J.M.; Weintraub, H. Cis-acting negative control of DNA replication in eukaryotic cells. Cell 1988, 52, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.M.; Cohen, S.N. Bovine papilloma virus plasmids replicate randomly in mouse fibroblasts throughout S phase of the cell cycle. Cell 1987, 50, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Ravnan, J.-B.; Gilbert, D.M.; Ten Hagen, K.G.; Cohen, S.N. Random-choice replication of extrachromosomal bovine papillomavirus (BPV) molecules in heterogeneous, clonally derived BPV-infected cell lines. J. Virol. 1992, 66, 6946–6952. [Google Scholar] [PubMed]
- Hoffmann, R.; Hirt, B.; Bechtold, V.; Beard, P.; Raj, K. Different modes of human papillomavirus DNA replication during maintenance. J. Virol. 2006, 80, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Moody, C.; Laimins, L.A.; Archambault, J. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J. Virol. 2010, 84, 11747–11760. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Lin, B.Y.; Deng, W.; Broker, T.R.; Chow, L.T. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J. Virol. 2007, 81, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- Straub, E.; Dreer, M.; Fertey, J.; Iftner, T.; Stubenrauch, F. The viral E8^E2C repressor limits productive replication of human papillomavirus 16. J. Virol. 2014, 88, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.; Laimins, L.A. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J. Virol. 2013, 87, 6037–6043. [Google Scholar] [CrossRef] [PubMed]
- Penrose, K.J.; McBride, A.A. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 2000, 74, 6031–6038. [Google Scholar] [CrossRef] [PubMed]
- Skiadopoulos, M.H.; McBride, A.A. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J. Virol. 1998, 72, 2079–2088. [Google Scholar] [PubMed]
- Ilves, I.; Kivi, S.; Ustav, M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which is mediated by the viral E2 protein and its binding sites. J. Virol. 1999, 73, 4404–4412. [Google Scholar] [PubMed]
- Bastien, N.; McBride, A.A. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology 2000, 270, 124–134. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Oliveira, J.G.; McPhillips, M.G. Partitioning viral genomes in mitosis: Same idea, different targets. Cell Cycle 2006, 5, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.G.; Colf, L.A.; McBride, A.A. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Kwon, D.; McBride, A.A. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J. Virol. 2009, 83, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Helfer, C.M.; Yan, J.; You, J. The cellular bromodomain protein Brd4 has multiple functions in E2-mediated papillomavirus transcription activation. Viruses 2014, 6, 3228–3249. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Anderson, D.E.; van Doorslaer, K.; McBride, A.A. A Proteomic approach to discover and compare interacting partners of Papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics 2015. [Google Scholar] [CrossRef]
- McBride, A.A. The Papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Singer, D.S. Two faces of brd4: Mitotic bookmark and transcriptional lynchpin. Transcription 2013, 4, 13–17. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Jang, M.K. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 2013, 5, 1374–1394. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.K.; McPhillips, M.G.; Ozato, K.; McBride, A.A. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 2005, 79, 4806–4818. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Ozato, K.; McBride, A.A. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 2005, 79, 8920–8932. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.R.; Ottinger, M.; You, J.; Howley, P.M. Brd4 independent transcriptional repression function of the papillomavirus E2 proteins. J. Virol. 2007, 81, 9612–9622. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Helfer, C.M.; Wang, R.; You, J. Analysis of the papillomavirus E2 and bromodomain protein Brd4 interaction using bimolecular fluorescence complementation. PLoS ONE 2013, 8, e77994. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-W.; Liu, W.-C.; Liao, K.-Y.; Tsao, Y.-P.; Hsu, P.-H.; Chen, S.-L. Phosphorylation of HPV-16 E2 at Serine 243 Enables Binding to Brd4 and Mitotic Chromosomes. PLoS ONE 2014, 9, e110882. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Colbert, A.M.; Laimins, L.A. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J. Virol. 1998, 72, 8115–8123. [Google Scholar] [PubMed]
- Senechal, H.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 2007, 358, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.Y.; Chiang, C.M.; McBride, A.A. Brd4 Is Displaced from HPV Replication Factories as They Expand and Amplify Viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2 mediated DNA replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed]
- Floyd, S.R.; Pacold, M.E.; Huang, Q.; Clarke, S.M.; Lam, F.C.; Cannell, I.G.; Bryson, B.D.; Rameseder, J.; Lee, M.J.; Blake, E.J.; et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 2013, 498, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Bedell, M.A.; Hudson, J.B.; Golub, T.R.; Turyk, M.E.; Hosken, M.; Wilbanks, G.D.; Laimins, L.A. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 1991, 65, 2254–2260. [Google Scholar] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Burnett, S.; Zabielski, J.; Moreno-Lopez, J.; Pettersson, U. Evidence for multiple vegetative DNA replication origins and alternative replication mechanisms of bovine papillomavirus type 1. J. Mol. Biol. 1989, 206, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Mutlu, E.; Sharma, V.; Collins, L.; Bodnar, W.; Yu, R.; Lai, Y.; Moeller, B.; Lu, K.; Swenberg, J. The endogenous exposome. DNA Repair 2014, 19, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Usui, T.; Petrini, J.H. Taking the time to make important decisions: The checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 2009, 8, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Chaurushiya, M.S.; Weitzman, M.D. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair 2009, 8, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.A. Viruses and the DNA Damage Response: Activation and Antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef]
- Sowd, G.A.; Li, N.Y.; Fanning, E. ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog. 2013, 9, e1003283. [Google Scholar] [CrossRef] [PubMed]
- Sowd, G.A.; Mody, D.; Eggold, J.; Cortez, D.; Friedman, K.L.; Fanning, E. SV40 utilizes ATM kinase activity to prevent non-homologous end joining of broken viral DNA replication products. PLoS Pathog. 2014, 10, e1004536. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-Dependent DNA Damage Response at the Human Papillomavirus 18 Replication Centers during the Initial Amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Kanginakudru, S.; DeSmet, M.; Thomas, Y.; Morgan, I.M.; Androphy, E.J. Levels of the E2 interacting protein TopBP1 modulate papillomavirus maintenance stage replication. Virology 2015, 478, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013, 9, e1003321. [Google Scholar] [CrossRef] [PubMed]
- Swindle, C.S.; Zou, N.; van Tine, B.A.; Shaw, G.M.; Engler, J.A.; Chow, L.T. Human papillomavirus DNA replication compartments in a transient DNA replication system. J. Virol. 1999, 73, 1001–1009. [Google Scholar] [PubMed]
- Stepp, W.H.; Meyers, J.M.; McBride, A.A. Sp100 provides intrinsic immunity against human papillomavirus infection. MBio 2013, 4, e00845–13. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol. 2006, 8, 365–374. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Fisk, J.C.; Dornan, E.S.; Donaldson, M.M.; Melendy, T.; Morgan, I.M. Human papillomavirus E1 and E2 mediated DNA replication is not arrested by DNA damage signalling. Virology 2010, 406, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, L.D.; Rivera Cardona, J.; Lambert, P.F. Inactivation of p53 rescues the maintenance of high risk HPV DNA genomes deficient in expression of E6. PLoS Pathog. 2013, 9, e1003717. [Google Scholar] [CrossRef] [PubMed]
- Lace, M.J.; Turek, L.P.; Anson, J.R.; Haugen, T.H. Analyzing the Human Papillomavirus (HPV) Life Cycle in Primary Keratinocytes with a Quantitative Colony-Forming Assay. Curr. Protoc. Microbiol. 2014, 33, 14b.2.1–14b.2.13. [Google Scholar] [PubMed]
- Leight, E.R.; Sugden, B. Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol. Cell. Biol. 2001, 21, 4149–4161. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA damage repair genes controlling human papillomavirus (HPV) episome levels under conditions of stability and extreme instability. PLoS ONE 2013, 8, e75406. [Google Scholar] [CrossRef] [PubMed]
- E, X.; Pickering, M.T.; Debatis, M.; Castillo, J.; Lagadinos, A.; Wang, S.; Lu, S.; Kowalik, T.F. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011, 7, e1001342. [Google Scholar] [CrossRef] [PubMed]
- Rogoff, H.A.; Pickering, M.T.; Frame, F.M.; Debatis, M.E.; Sanchez, Y.; Jones, S.; Kowalik, T.F. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol. Cell. Biol. 2004, 24, 2968–2977. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Park, R.B.; Androphy, E.J. Genetic analysis of high-risk e6 in episomal maintenance of human papillomavirus genomes in primary human keratinocytes. J. Virol. 2002, 76, 11359–11364. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Vande Pol, S.B. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal human papillomavirus genome maintenance. J. Virol. 2014, 88, 3027–3030. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.J.; Laimins, L.A. Differentiation-induced changes in promoter usage for transcripts encoding the human papillomavirus type 31 replication protein E1. Virology 1999, 257, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. The JAK-STAT Transcriptional Regulator, STAT-5, Activates the ATM DNA Damage Pathway to Induce HPV 31 Genome Amplification upon Epithelial Differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [PubMed]
- Kho, E.Y.; Wang, H.K.; Banerjee, N.S.; Broker, T.R.; Chow, L.T. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc. Natl. Acad. Sci. USA 2013, 110, 7542–7549. [Google Scholar] [CrossRef] [PubMed]
- Lepik, D.; Ilves, I.; Kristjuhan, A.; Maimets, T.; Ustav, M. p53 protein is a suppressor of papillomavirus DNA amplificational replication. J. Virol. 1998, 72, 6822–6831. [Google Scholar]
- Ilves, I.; Kadaja, M.; Ustav, M. Two separate replication modes of the bovine papillomavirus BPV1 origin of replication that have different sensitivity to p53. Virus Res. 2003, 96, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Akgul, B.; Karle, P.; Adam, M.; Fuchs, P.G.; Pfister, H.J. Dual role of tumor suppressor p53 in regulation of DNA replication and oncogene E6-promoter activity of epidermodysplasia verruciformis-associated human papillomavirus type 8. Virology 2003, 308, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Kowalczyk, A.M.; Taylor, E.R.; Morgan, I.M.; Gaston, K. P53 represses human papillomavirus type 16 DNA replication via the viral E2 protein. Virol. J. 2008, 5, e5. [Google Scholar] [CrossRef]
- Dasgupta, S.; Zabielski, J.; Simonsson, M.; Burnett, S. Rolling-circle replication of a high-copy BPV-1 plasmid. J. Mol. Biol. 1992, 228, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Auborn, K.J.; Little, R.D.; Platt, T.H.; Vaccariello, M.A.; Schildkraut, C.L. Replicative intermediates of human papillomavirus type 11 in laryngeal papillomas: Site of replication initiation and direction of replication. Proc. Natl. Acad. Sci. USA 1994, 91, 7340–7344. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, J.R.; Lipkin-Moore, Z.; Sheu, Y.J.; Stillman, B.; Burgers, P.M.; Haber, J.E. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 2010, 24, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Galloway, D.A. Beta human papillomavirus E6 expression inhibits stabilization of p53 and increases tolerance of genomic instability. J. Virol. 2014, 88, 6112–6127. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. beta-HPV 5 and 8 E6 Disrupt Homology Dependent Double Strand Break Repair by Attenuating BRCA1 and BRCA2 Expression and Foci Formation. PLoS Pathog. 2015, 11, e1004687. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Storey, A. E6 proteins from diverse cutaneous HPV types inhibit apoptosis in response to UV damage. Oncogene 2000, 19, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Storey, A. Repair of UV-induced thymine dimers is compromised in cells expressing the E6 protein from human papillomaviruses types 5 and 18. Br. J. Cancer 2004, 90, 2203–2209. [Google Scholar] [PubMed]
- Holloway, A.; Simmonds, M.; Azad, A.; Fox, J.L.; Storey, A. Resistance to UV-induced apoptosis by beta-HPV5 E6 involves targeting of activated BAK for proteolysis by recruitment of the HERC1 ubiquitin ligase. Int. J. Cancer J. Int. Cancer 2015, 136, 2831–2843. [Google Scholar] [CrossRef]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Myers, S.L.; Gostout, B.S.; Smith, D.I. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 2003, 22, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.P.; Friedman, C.L.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 1992, 5, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2013. [Google Scholar] [CrossRef]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses 2015, 7, 2450-2469. https://doi.org/10.3390/v7052450
McKinney CC, Hussmann KL, McBride AA. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses. 2015; 7(5):2450-2469. https://doi.org/10.3390/v7052450
Chicago/Turabian StyleMcKinney, Caleb C., Katherine L. Hussmann, and Alison A. McBride. 2015. "The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle" Viruses 7, no. 5: 2450-2469. https://doi.org/10.3390/v7052450
APA StyleMcKinney, C. C., Hussmann, K. L., & McBride, A. A. (2015). The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses, 7(5), 2450-2469. https://doi.org/10.3390/v7052450