
The Dual Role of Exosomes in Hepatitis A and C Virus Transmission and Viral Immune Activation

{kind=link}
Abstract
:1. Introduction
2. Exosomes Emerge as Important Players in Viral Pathogenesis
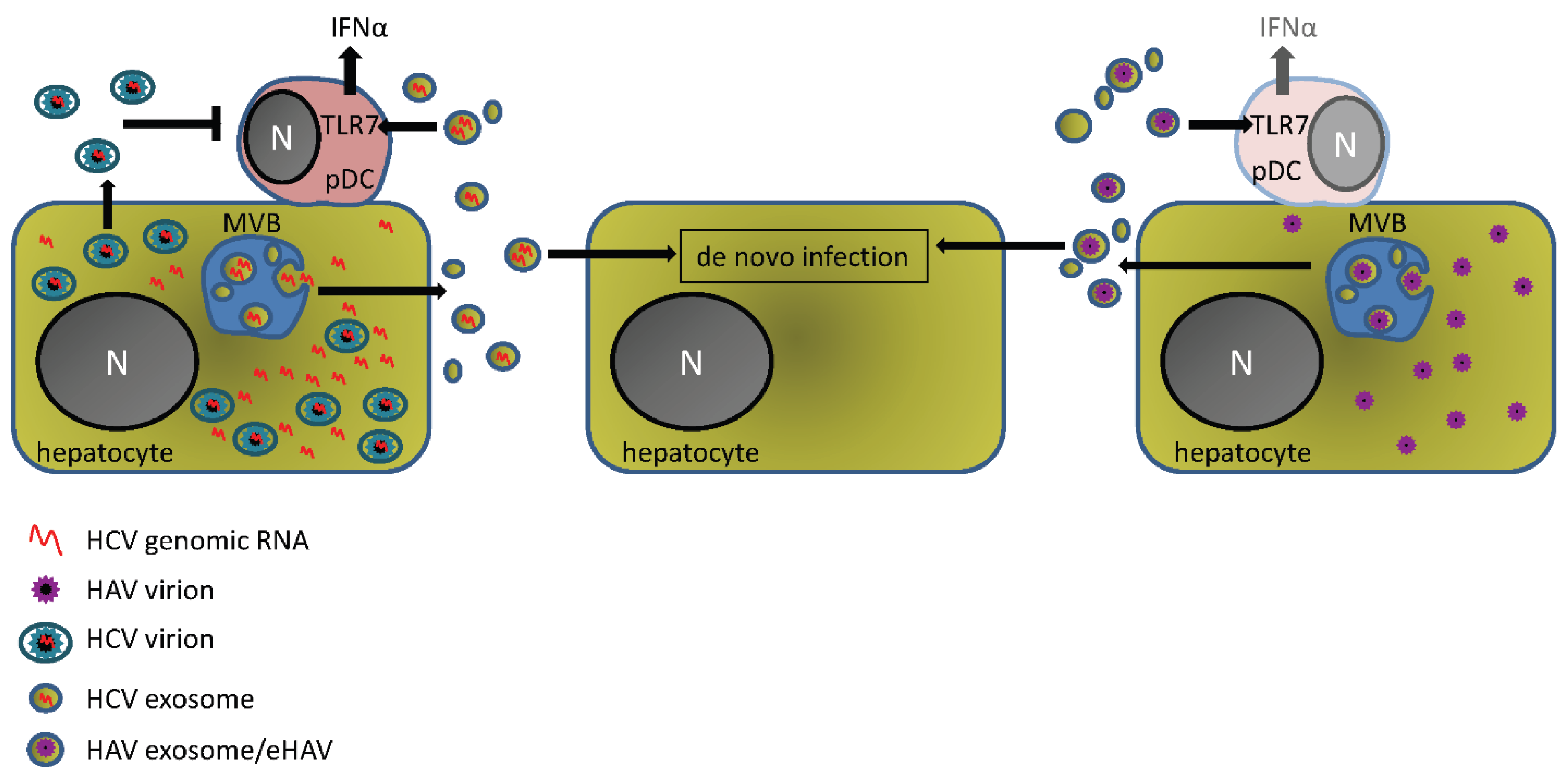
3. Exosomal Transport of Infectious Viral Agents and Activation of the Interferon Response
4. Viral Exosomes as Immune Evasive Infectious Particles
5. Discussion
Acknowledgments
Conflicts of Interest
References
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Harding, C.; Heuser, J.; Stahl, P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: Demonstration of a pathway for receptor shedding. Eur. J. Cell Biol. 1984, 35, 256–263. [Google Scholar] [PubMed]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Stenmark, H.; Emr, S.D. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harbor Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Bao, X.; Casola, A. Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses 2015, 7, 3204–3225. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [PubMed]
- Conde-Vancells, J.; Rodriguez-Suarez, E.; Embade, N.; Gil, D.; Matthiesen, R.; Valle, M.; Elortza, F.; Lu, S.C.; Mato, J.M.; Falcon-Perez, J.M. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J. Proteome Res. 2008, 7, 5157–5166. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- Negro, F.; Alberti, A. The global health burden of hepatitis C virus infection. Liver Int. 2011, 31, S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Owen, D.M.; Li, K.; Erickson, A.K.; Johnson, C.L.; Fish, P.M.; Carney, D.S.; Wang, T.; Ishida, H.; Yoneyama, M.; et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 6001–6006. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhong, J.; Chisari, F.V. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8499–8504. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Serti, E.; Eke, O.; Muchmore, B.; Prokunina-Olsson, L.; Capone, S.; Folgori, A.; Rehermann, B. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology 2012, 56, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.E.; Giugliano, S.; Schnell, G.; Cheng, L.; Leahy, K.F.; Golden-Mason, L.; Gale, M., Jr.; Rosen, H.R. Hepatitis C virus pathogen associated molecular pattern (PAMP) triggers production of lambda-interferons by human plasmacytoid dendritic cells. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Giugliano, S.; Kriss, M.; Golden-Mason, L.; Dobrinskikh, E.; Stone, A.E.; Soto-Gutierrez, A.; Mitchell, A.; Khetani, S.R.; Yamane, D.; Stoddard, M.; et al. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 2015, 148, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kodys, K.; Li, K.; Szabo, G. Human type 2 myeloid dendritic cells produce interferon-λ and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology 2013, 144, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Boyd, B.; Chisari, F.V. Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J. Virol. 2015, 89, 2956–2961. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, X.; Yu, Q.; He, J.J. Exosome-associated hepatitis C virus in cell cultures and patient plasma. Biochem. Biophys. Res. Commun. 2014, 455, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Shiina, M.; Rehermann, B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology 2008, 47, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Kanto, T.; Inoue, M.; Miyatake, H.; Sato, A.; Sakakibara, M.; Yakushijin, T.; Oki, C.; Itose, I.; Hiramatsu, N.; Takehara, T.; et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J. Infect. Dis. 2004, 190, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Florentin, J.; Aouar, B.; Dental, C.; Thumann, C.; Firaguay, G.; Gondois-Rey, F.; Soumelis, V.; Baumert, T.F.; Nunès, J.A.; Olive, D.; et al. HCV glycoprotein E2 is a novel BDCA-2 ligand and acts as an inhibitor of IFN production by plasmacytoid dendritic cells. Blood 2012, 120, 4544–4551. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Feng, Z.; Yamane, D.; Liang, Y.; Lanford, R.E.; Li, K.; Lemon, S.M. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Lemon, S.M. Hepatitis A virus: From discovery to vaccines. Hepatology 2006, 43, S164–S172. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M. Type A viral hepatitis. New developments in an old disease. N. Engl. J. Med. 1985, 313, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Lanforda, R.E.; Fengc, Z.; Chaveza, D.; Guerraa, B.; Braskyb, K.M.; Zhoud, Y.; Yamanec, D.; Perelsone, A.S.; Walkerd, C.M.; Lemonc, S.M. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 11223–11228. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Victor, J.C.; Monto, A.S.; Surdina, T.Y.; Suleimenova, S.Z.; Vaughan, G.; Nainan, O.V.; Favorov, M.O.; Margolis, H.S.; Bell, B.P. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N. Engl. J. Med. 2007, 357, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, Y.; McKnight, K.L.; Hensley, L.; Lanford, R.E.; Walker, C.M.; Lemon, S.M. Human pDCs preferentially sense enveloped hepatitis A virions. J. Clin. Invest. 2015, 125, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Date, T.; Miyamoto, M.; Furusaka, A.; Tokushige, K.; Mizokami, M.; Wakita, T. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 2003, 125, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Lohmann, V.; Kaul, A.; Krieger, N.; Rinck, G.; Rutter, G.; Strand, D.; Bartenschlager, R. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 2002, 76, 4008–4021. [Google Scholar] [CrossRef] [PubMed]
- Assil, S.; Webster, B.; Dreux, M. Regulation of the host antiviral state by intercellular communications. Viruses 2015, 7, 4707–4733. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Shiina, M.; Tanaka, N.; Nakano, T.; Yamamoto, A.; Kondo, Y.; Kakazu, E.; Inoue, J.; Fukushima, K.; Sano, K.; et al. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 2012, 422, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Alenquer, M.; Amorim, M.J. Exosome biogenesis, regulation, and function in viral infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xiao, Z.D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [PubMed]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Mörgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Barres, C.; Blanc, L.; Bette-Bobillo, P.; André, S.; Mamoun, R.; Gabius, H.J.; Vidal, M. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T.; et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 2015. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Kobayashi, N.B.; Takatani-Nakase, T.; Yoshida, T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Masciopinto, F.; Giovani, C.; Campagnoli, S.; Galli-Stampino, L.; Colombatto, P.; Brunetto, M.; Yen, T.S.; Houghton, M.; Pileri, P.; Abrignani, S. Association of hepatitis C virus envelope proteins with exosomes. Eur. J. Immunol. 2004, 34, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Welsch, C.; Hensley, L.; Jangra, R.K.; Lemon, S.M. Base pairing between hepatitis C virus RNA and microRNA 122 3' of its seed sequence is essential for genome stabilization and production of infectious virus. J. Virol. 2012, 86, 7372–7383. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Nishimura, Y.; Ichimura, T.; Suzuki, K.; Miyamura, T.; Suzuki, T.; Moriishi, K.; Matsuura, Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006, 25, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Zhang, C.; Huys, A.; Richardson, C.D. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J. Virol. 2011, 85, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Schutz, S.; Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008, 4, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 2010, 84, 6615–6625. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.F.; Welch, M.; Schreiber, R.D.; de la Torre, J.D.; Oldstone, M.B.A. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, P.M.; Wherry, E.J. Immunology. An interferon paradox. Science 2013, 340, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Takahashi, K.; Boyd, B.; Whitten-Bauer, C.; Ngo, N.; de la Torre, J.C.; Chisari, F.V. Human plasmacytoid dendritic cells sense lymphocytic choriomeningitis virus-infected cells in vitro. J. Virol. 2014, 88, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.; Pothlichet, J.; Schilte, C.; Chaperot, L.; Plumas, J.; Randall, R.E.; Si-Tahar, M.; et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longatti, A. The Dual Role of Exosomes in Hepatitis A and C Virus Transmission and Viral Immune Activation. Viruses 2015, 7, 6707-6715. https://doi.org/10.3390/v7122967
Longatti A. The Dual Role of Exosomes in Hepatitis A and C Virus Transmission and Viral Immune Activation. Viruses. 2015; 7(12):6707-6715. https://doi.org/10.3390/v7122967
Chicago/Turabian StyleLongatti, Andrea. 2015. "The Dual Role of Exosomes in Hepatitis A and C Virus Transmission and Viral Immune Activation" Viruses 7, no. 12: 6707-6715. https://doi.org/10.3390/v7122967
APA StyleLongatti, A. (2015). The Dual Role of Exosomes in Hepatitis A and C Virus Transmission and Viral Immune Activation. Viruses, 7(12), 6707-6715. https://doi.org/10.3390/v7122967