
Harnessing the Power of Onco-Immunotherapy with Checkpoint Inhibitors

Abstract
:
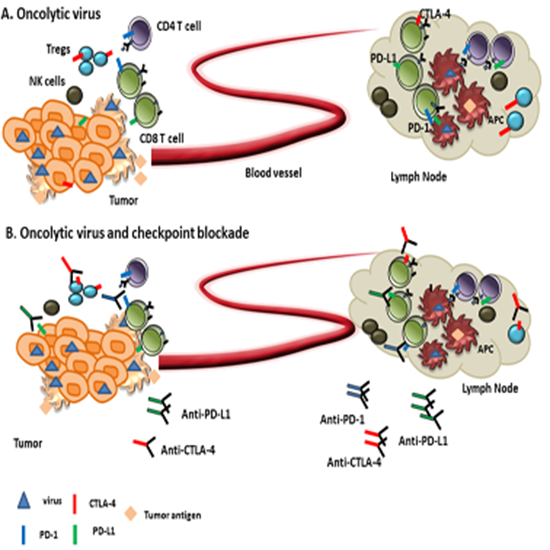{kind=link}
1. Introduction
2. Onco-Immunotherapy
3. Mechanisms of Tumor-Mediated Immune Tolerance
4. CTLA-4 and PD-1 Checkpoint Molecules
5. Oncolytic Viruses with Anti-CTLA-4 Therapy
6. Viruses Engineered to Express Antibodies Targeting Checkpoint Molecules
7. Reovirus with Anti-PD-1 Therapy
8. Moving Forward
9. Conclusions
Author Contributions
Conflicts of Interest
References
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Todo, T. “Armed” oncolytic herpes simplex viruses for brain tumor therapy. Cell Adhes. Migr. 2008, 2, 208–213. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [PubMed]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273. [Google Scholar] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Dahm-Vicker, M.; Golgher, D.; Gallimore, A. CD25+ regulatory T cells and tumor immunity. Immunol. Lett. 2003, 85, 141–143. [Google Scholar] [CrossRef]
- Orentas, R.J.; Kohler, M.E.; Johnson, B.D. Suppression of anti-cancer immunity by regulatory T cells: Back to the future. Semin. Cancer Biol. 2006, 16, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; de Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Egen, J.G.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef]
- Darlington, P.J.; Baroja, M.L.; Chau, T.A.; Siu, E.; Ling, V.; Carreno, B.M.; Madrenas, J. Surface cytotoxic T lymphocyte-associated antigen 4 partitions within lipid rafts and relocates to the immunological synapse under conditions of inhibition of T cell activation. J. Exp. Med. 2002, 195, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L.; Mao, M.; Kobayashi, S.; Biery, M.; Burchard, J.; Cavet, G.; Gregson, B.P.; June, C.H.; Linsley, P.S. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 11790–11795. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Appleman, L.J.; van Puijenbroek, A.A.; Shu, K.M.; Nadler, L.M.; Boussiotis, V.A. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J. Immunol. 2002, 168, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular basis of T cell inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar] [CrossRef] [PubMed]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [PubMed]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune modulation for cancer therapy. Br. J. Cancer 2014, 111, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Van den Eertwegh, A.J.; Versluis, J.; van den Berg, H.P.; Santegoets, S.J.; van Moorselaar, R.J.; van der Sluis, T.M.; Gall, H.E.; Harding, T.C.; Jooss, K.; Lowy, I.; et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 509–517. [Google Scholar] [CrossRef]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.L.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. CII 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Hammers, H.J.; Infante, E.R.P.J.R.; Rini, B.I.; McDermott, D.F.; Ernstoff, M.; Voss, M.H.; Sharma, P.; Pal, S.K.; Razak, A.R.A.; Kollmannsberger, C.K.; et al. Expanded Cohort Results from CheckMate 016: A Phase I Study of Nivolumab in Combination with Ipilimumab in Metastatic Renal Cell Carcinoma (mRCC); ASCO: Chicago, IL, USA, 2015. [Google Scholar]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Woo, S.R.; Zha, Y.; Spaapen, R.; Zheng, Y.; Corrales, L.; Spranger, S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Piasecki, P.; Tiep, L.; Ponce, R.; Beers, C. Talilmogene Laherparepvec Increases the Anti-Tumor Efficacy of the Anti-PD-1 Immune Checkpoint Blockade; AACR: Philadelphia, PA, USA, 2015. [Google Scholar]
- Fiola, C.; Peeters, B.; Fournier, P.; Arnold, A.; Bucur, M.; Schirrmacher, V. Tumor selective replication of Newcastle disease virus: Association with defects of tumor cells in antiviral defence. Int. J. Cancer 2006, 119, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Palese, P. Oncolytic Newcastle disease virus for cancer therapy: Old challenges and new directions. Future Microbiol. 2012, 7, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; McFadden, G. Oncolytic Poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [PubMed]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Park, B.H.; Hwang, T.; Liu, T.C.; Sze, D.Y.; Kim, J.S.; Kwon, H.C.; Oh, S.Y.; Han, S.Y.; Yoon, J.H.; Hong, S.H.; et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: A phase I trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef]
- Barber, G.N. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004, 17, 516–527. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute, M.C. Viral Therapy in Treating Patient with Liver Cancer; National Library of Medicine: Bethesda, MD, USA, 2012. Available online: https://clinicaltrials.gov/show/NCT01628640 (accessed on 5 June 2015).
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Errington, F.; Pulido, J.; Galivo, F.; Thompson, J.; Wongthida, P.; Diaz, R.M.; Chong, H.; Ilett, E.; Chester, J.; et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat. Med. 2011, 17, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Pulido, J.; Kottke, T.; Thompson, J.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Rommelfanger, D.; Ilett, E.; Pease, L.; Pandha, H.; et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat. Biotechnol. 2012, 30, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Camino, V.; Rajani, K.; Kottke, T.; Rommelfanger-Konkol, D.; Zaidi, S.; Thompson, J.; Pulido, J.; Ilett, E.; Donnelly, O.; Selby, P.; et al. The profile of tumor antigens which can be targeted by immunotherapy depends upon the tumor's anatomical site. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Cockle, J.V.R.K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.; Shim, K.S.; Peterson, T.; Parney, I.; Short, S.; Selby, P.; et al. Combination Viroimmunotherapy with Checkpoint Inhibition to Treat Glioma, based on Location-Specific Tumor Profiling. Neuro Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Dispenzieri, A.; Galanis, E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr. Pharm. Biotechnol. 2012, 13, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute, M.C. Viral Therapy in Treating Patients with Recurrent Glioblastoma Multiforme; National Library of Medicine: Bethesda, MD, USA, 2006. Available online: https://clinicaltrials.gov/show/NCT00390299 (accessed on 5 June 2015).
- National Cancer Institute, M.C. MV-NIS or Investigator’s Choice Chemotherapy in Treating Patients with Ovarian, Fallopian, or Peritoneal Cancer; National Library of Medicine: Bethesda, MD, USA, 2015. Available online: https://clinicaltrials.gov/show/NCT02364713 (accessed on 10 June 2015).
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Errington, F.; Steele, L.; Prestwich, R.; Harrington, K.J.; Pandha, H.S.; Vidal, L.; de Bono, J.; Selby, P.; Coffey, M.; Vile, R.; et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J. Immunol. 2008, 180, 6018–6026. [Google Scholar] [CrossRef] [PubMed]
- Comins, C.; Spicer, J.; Protheroe, A.; Roulstone, V.; Twigger, K.; White, C.M.; Vile, R.; Melcher, A.; Coffey, M.C.; Mettinger, K.L.; et al. REO-10: A phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5564–5572. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Markovic, S.N.; Suman, V.J.; Nuovo, G.J.; Vile, R.G.; Kottke, T.J.; Nevala, W.K.; Thompson, M.A.; Lewis, J.E.; Rumilla, K.M.; et al. Phase II trial of intravenous administration of Reolysin((R)) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1998–2003. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol. Ther. J. Am. Soc. Gene Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol. Immunother. CII 2014, 63, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Ferrantini, M.; Capone, I.; Belardelli, F. Interferon-alpha and cancer: Mechanisms of action and new perspectives of clinical use. Biochimie 2007, 89, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA approves first-of-its-kind product for the treatment of melanoma. In FDA News Release; FDA: Silver Spring, MD, USA, 2015. [Google Scholar]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajani, K.R.; Vile, R.G. Harnessing the Power of Onco-Immunotherapy with Checkpoint Inhibitors. Viruses 2015, 7, 5889-5901. https://doi.org/10.3390/v7112914
Rajani KR, Vile RG. Harnessing the Power of Onco-Immunotherapy with Checkpoint Inhibitors. Viruses. 2015; 7(11):5889-5901. https://doi.org/10.3390/v7112914
Chicago/Turabian StyleRajani, Karishma R., and Richard G. Vile. 2015. "Harnessing the Power of Onco-Immunotherapy with Checkpoint Inhibitors" Viruses 7, no. 11: 5889-5901. https://doi.org/10.3390/v7112914
APA StyleRajani, K. R., & Vile, R. G. (2015). Harnessing the Power of Onco-Immunotherapy with Checkpoint Inhibitors. Viruses, 7(11), 5889-5901. https://doi.org/10.3390/v7112914