
Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications



Abstract

1. Adenovirus Immunogenicity
1.1. Introduction
1.2. Tissue Tropism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Adenovirus Classification and Tropism | |||
|---|---|---|---|
| Species | Serotypes | Receptors | Tropism |
| A | 12, 18, 31 | hCAR | Cryptic (enteric, respiratory) |
| B1 | 16, 21, 35, 50 | CD46 [19], CD80, CD86 | Respiratory, ocular |
| B2 | 3, 7, 14, 34, 35 | DSG-2 [18], CD80, CD86 | Renal, ocular, respiratory |
| B1/2 | 11 | CD46, DSG-2 [18] | Ocular, respiratory |
| C | 1, 2, 5, 6 | hCAR [20], HSPG, VCAM-1, SR, MHC1-α2 | Respiratory, ocular, lymphoid |
| D | 8‒10, 13, 15, 17, 19, 20, 22‒30, 32, 33, 36‒39, 42‒49, 51, 53 | SA [21], CD46 [22,23], hCAR [22], GD1a glycan [24] | Ocular, Enteric |
| E | 4 | hCAR | Ocular, respiratory |
| F | 40, 41 | hCAR | Enteric |
| G | 52 | nd | Enteric |
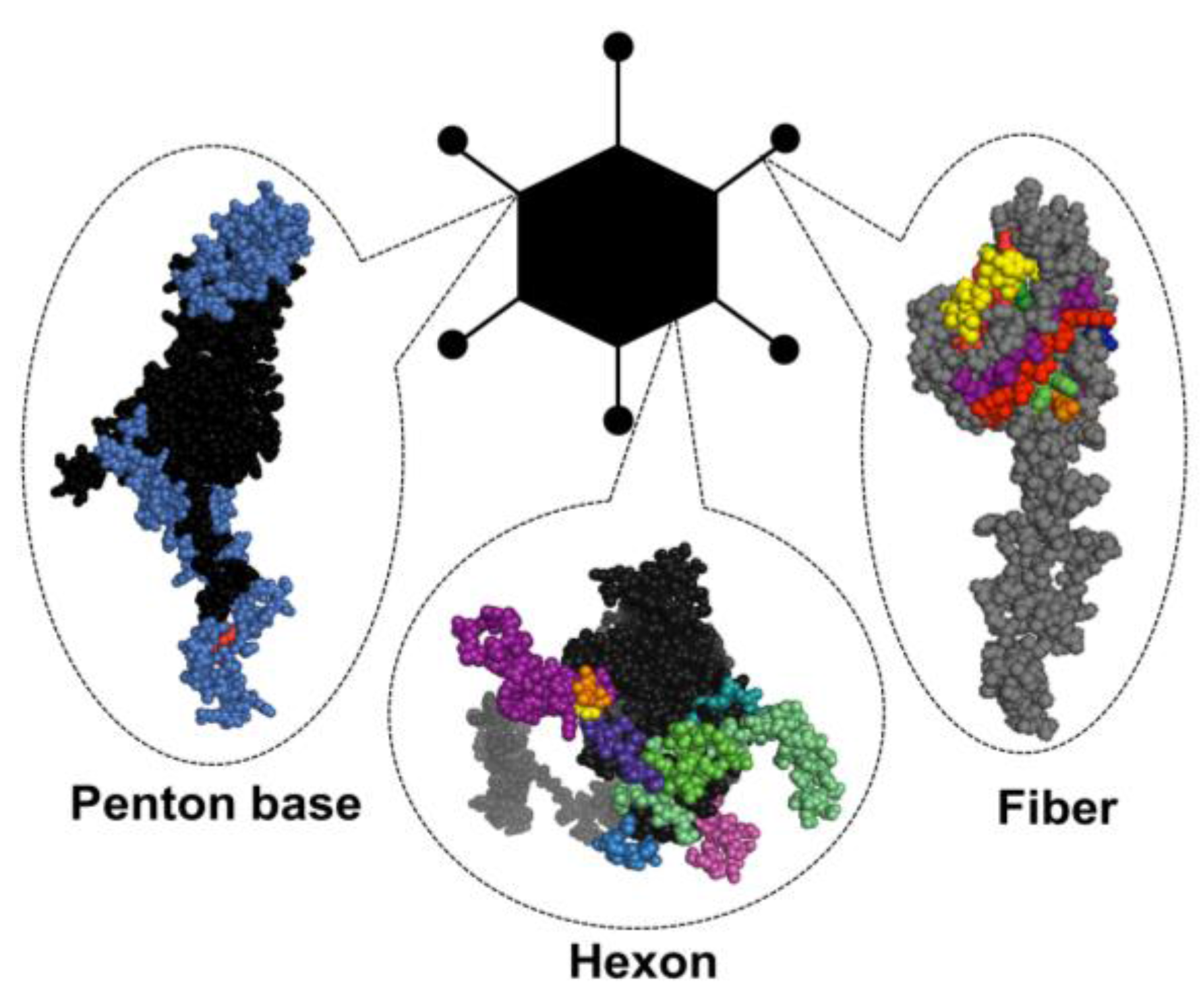
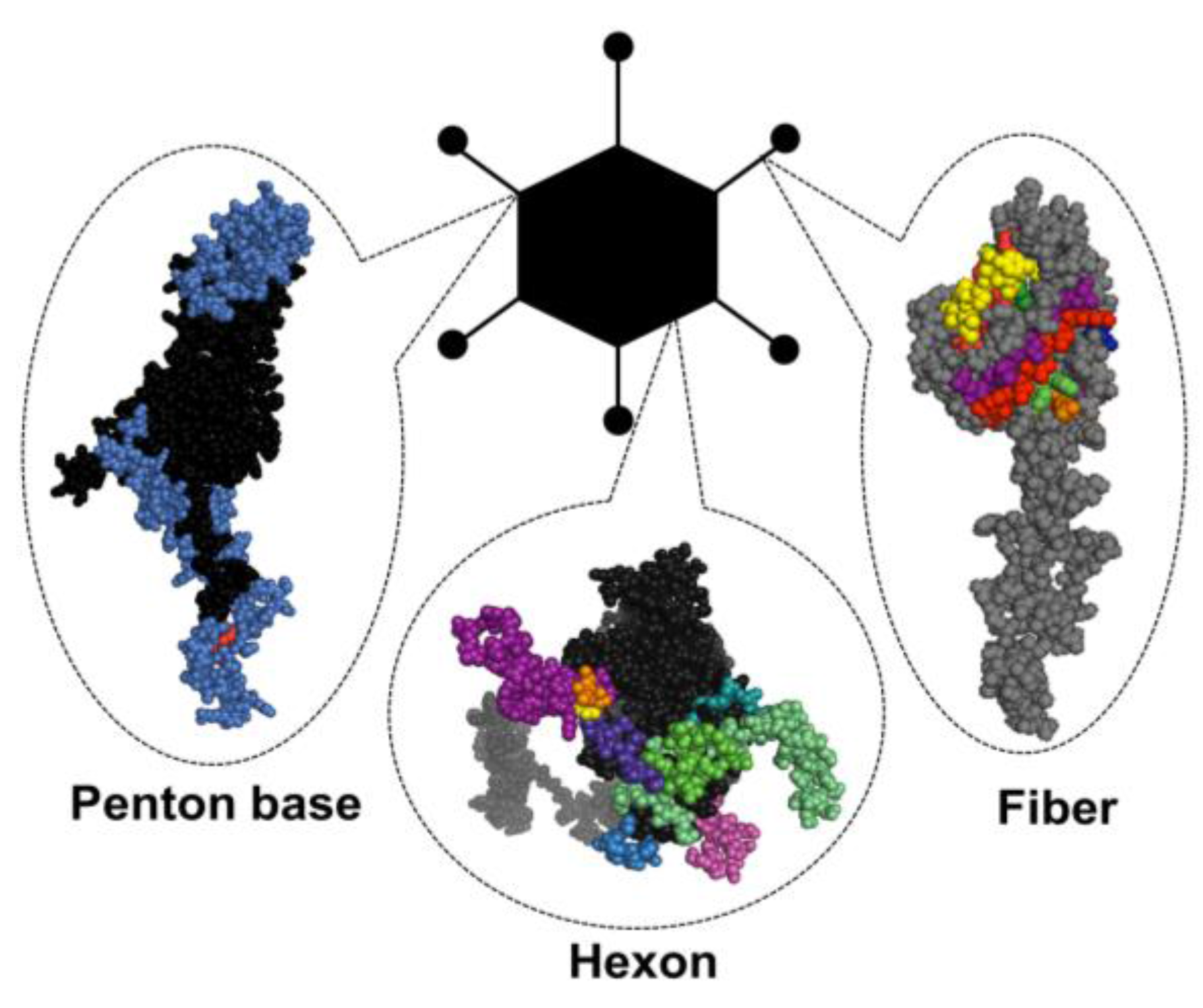
1.3. Structural Basis of Immunogenicity
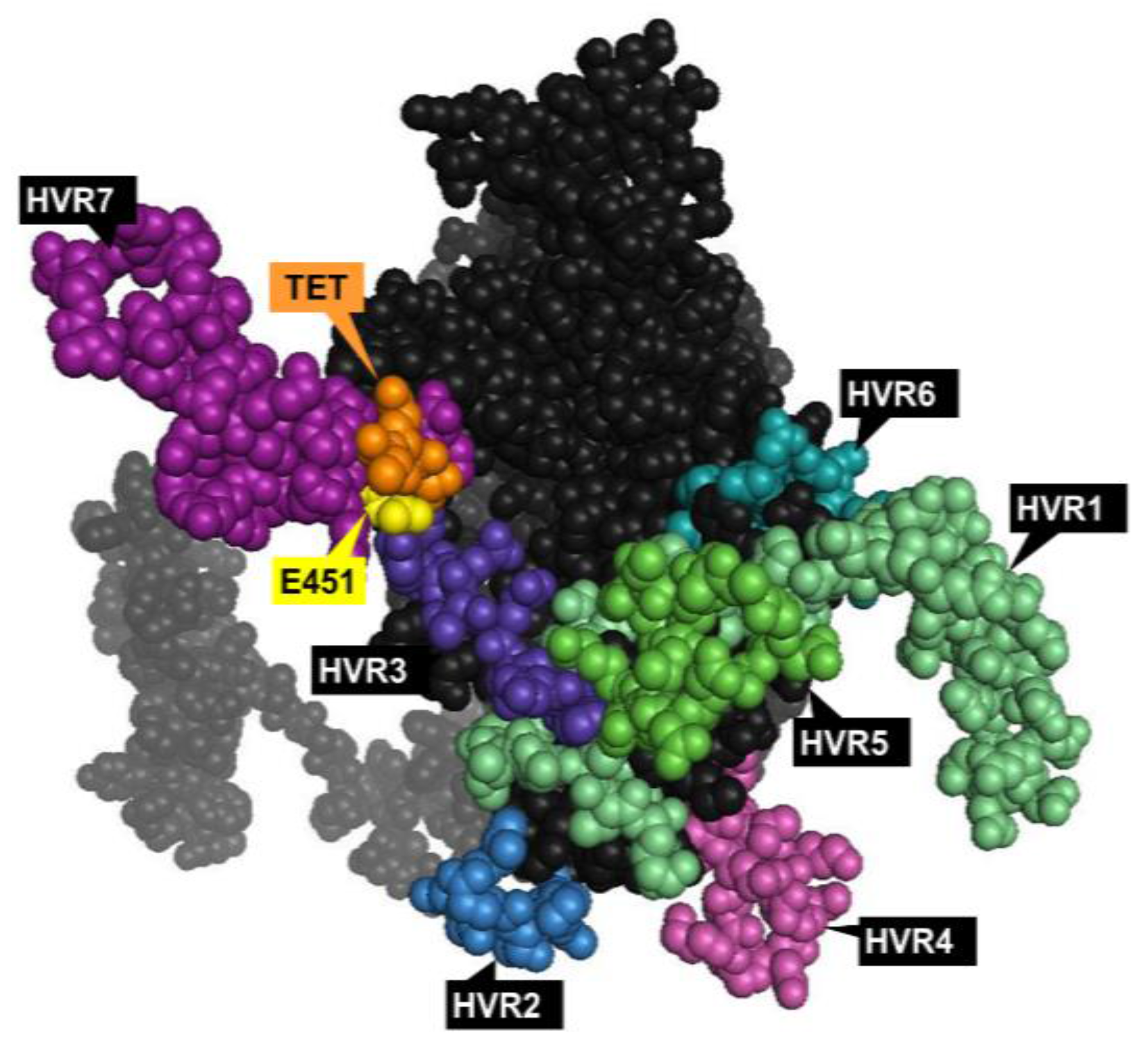
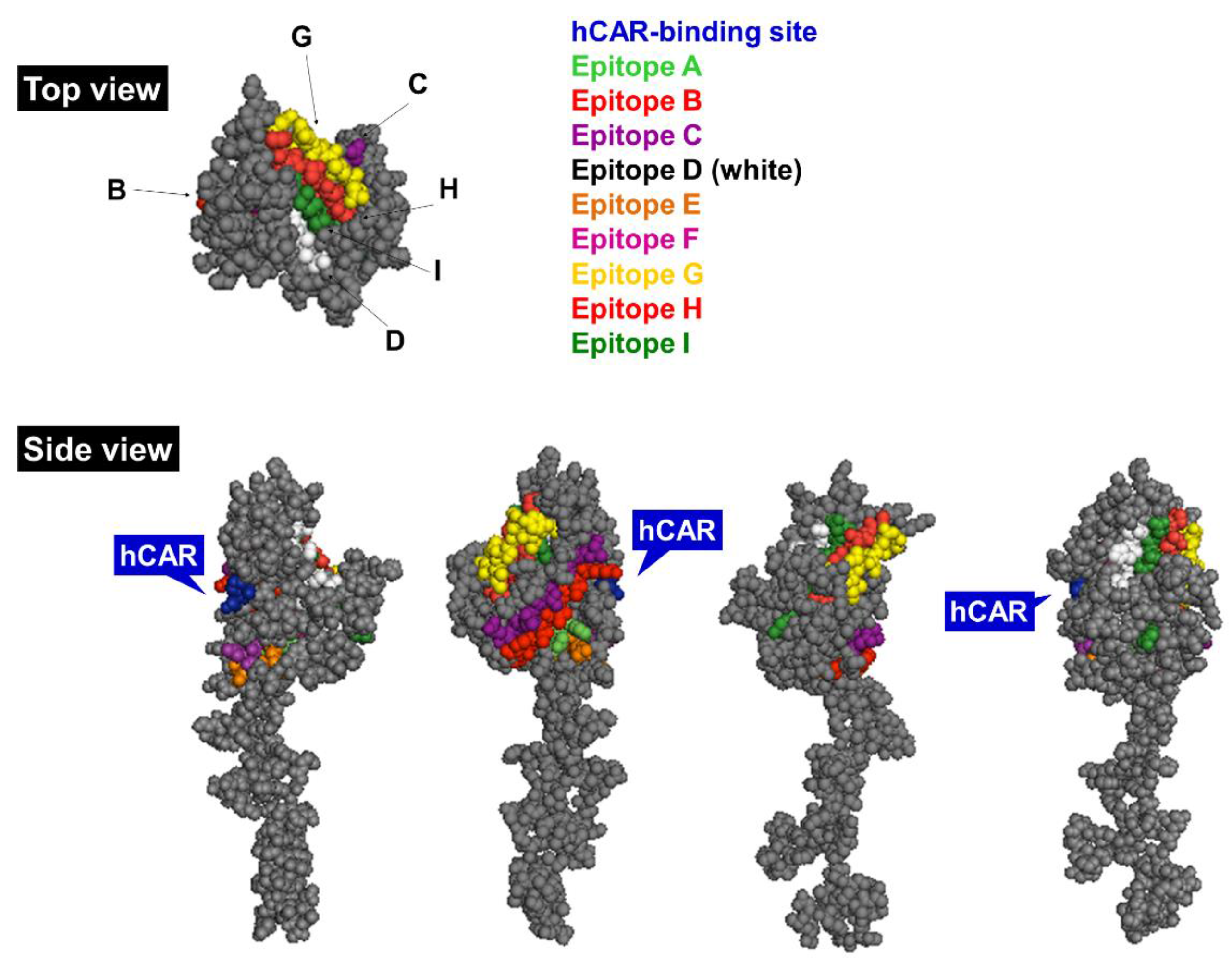
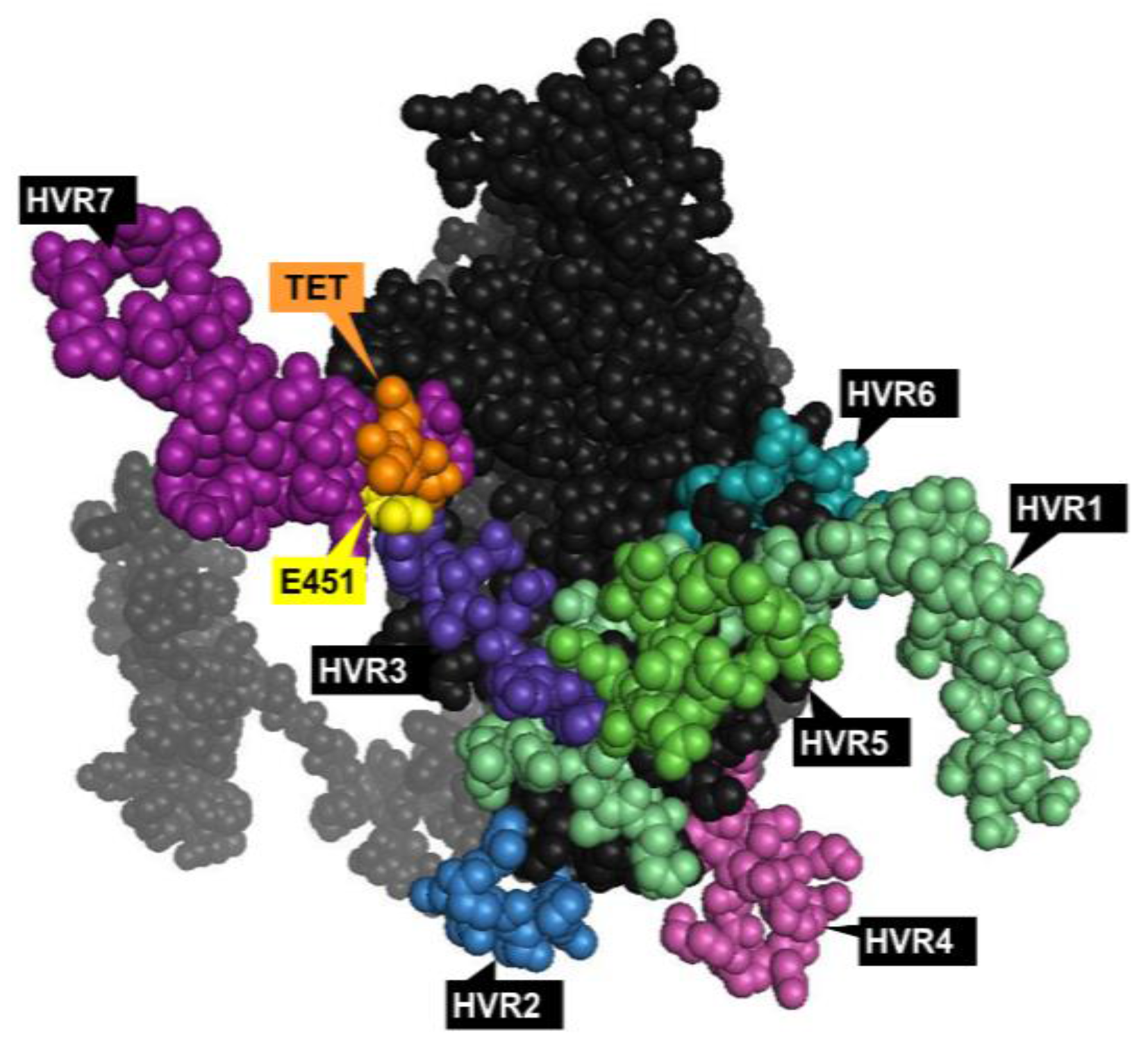
1.3.1. Hexon
1.3.2. Fiber
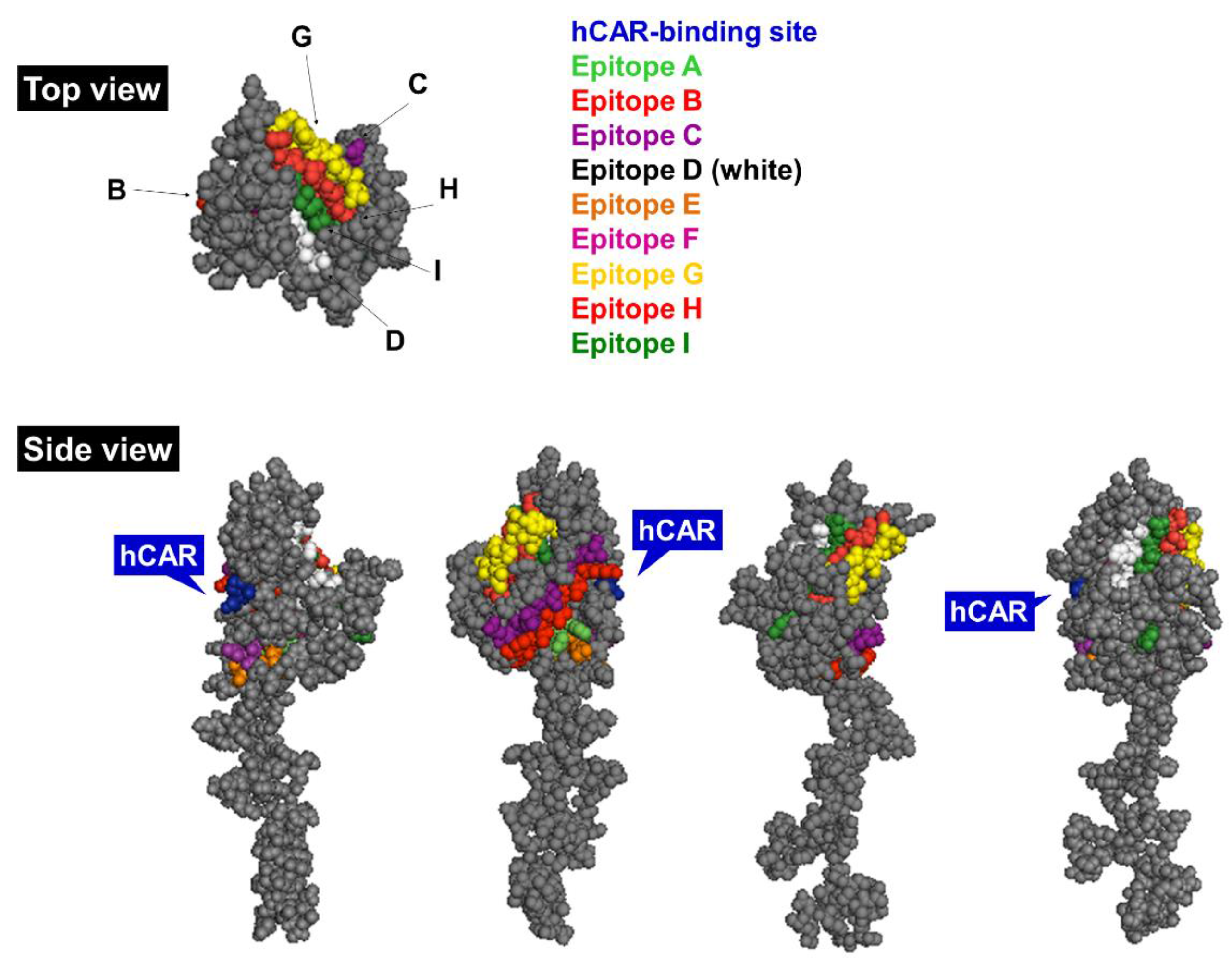
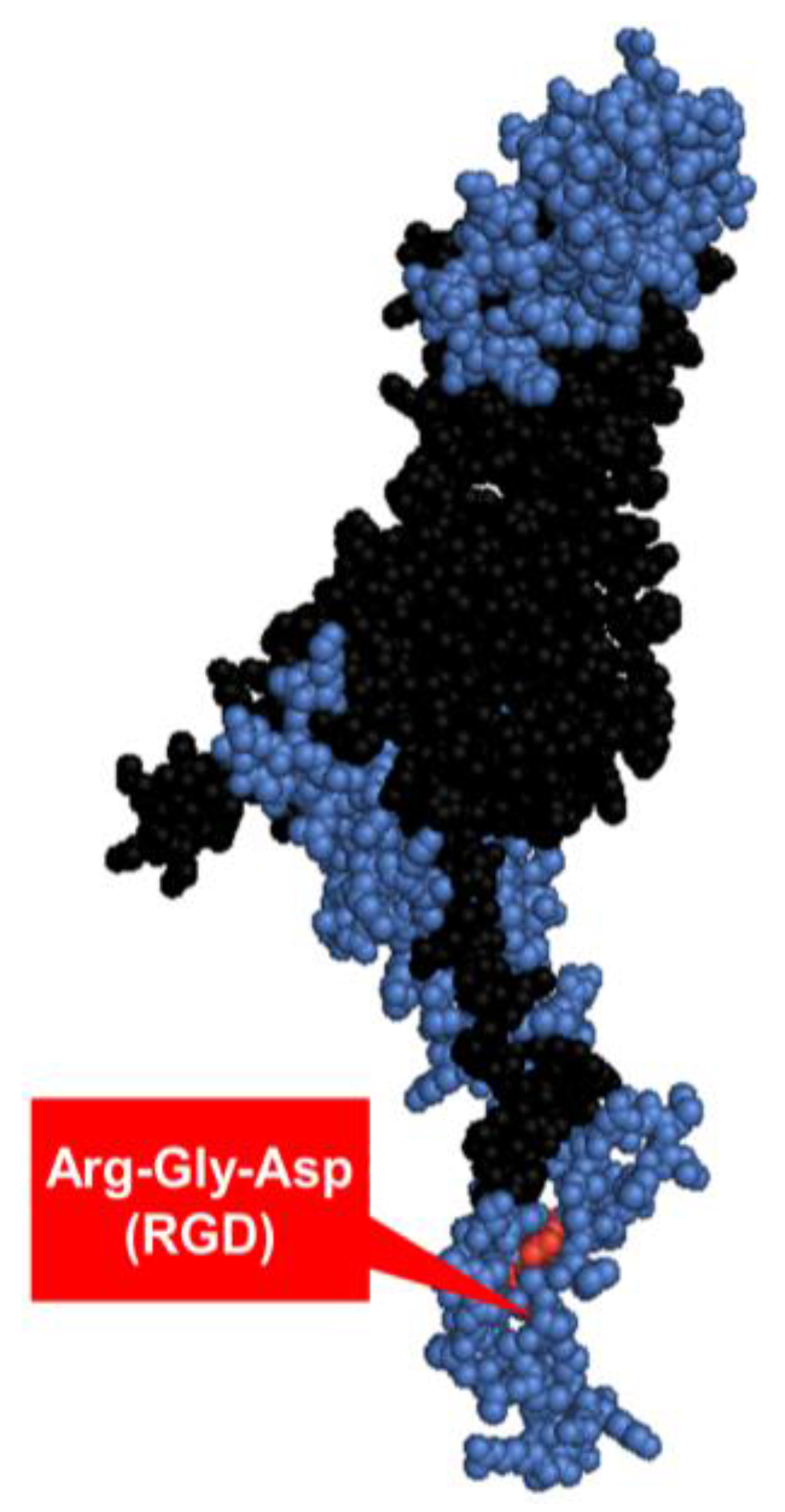
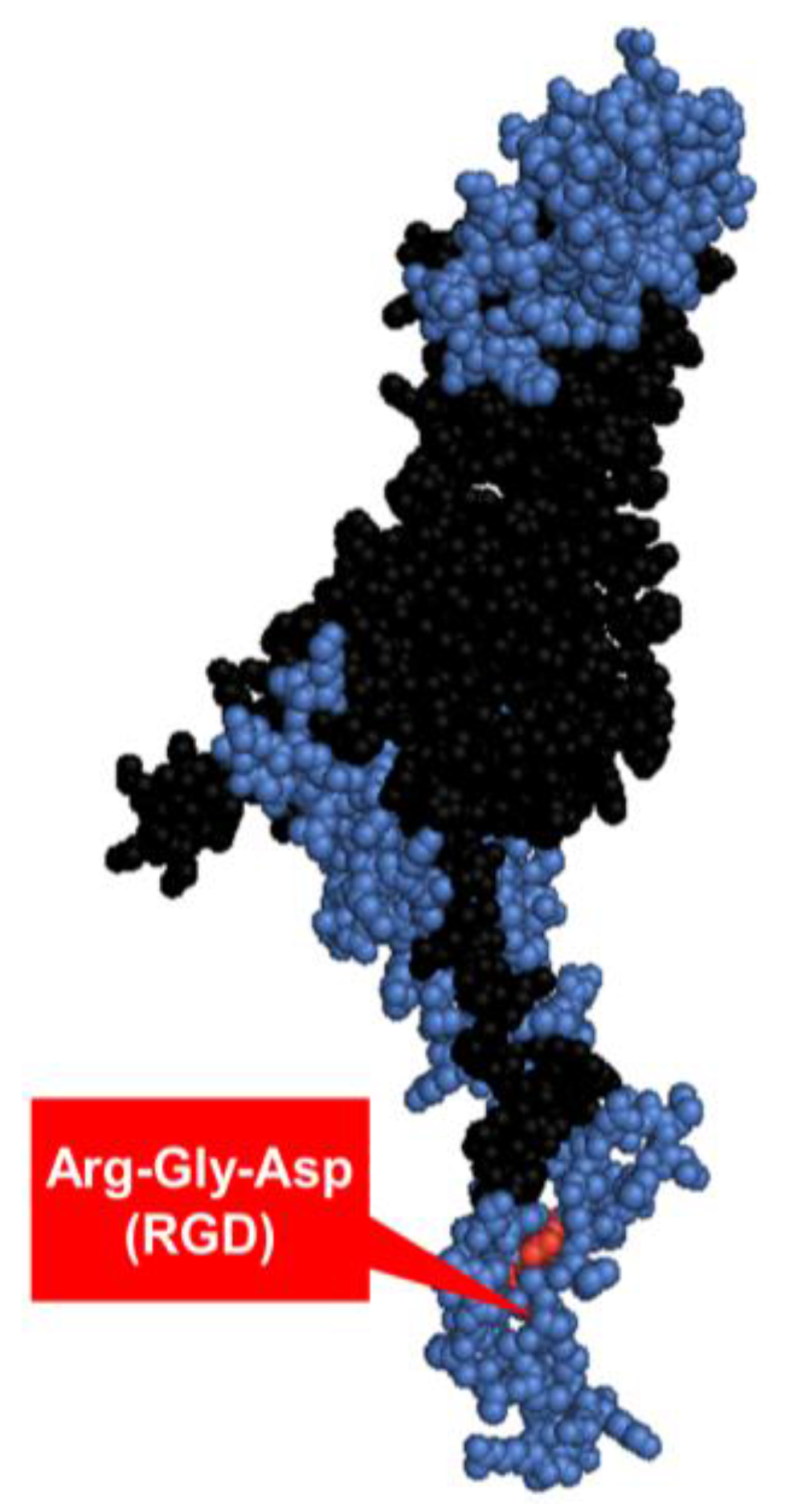
1.3.3. Penton base
1.4. Innate Immune Responses
1.5. Adaptive Immune Responses
1.6. Natural vs. Induced Immunity
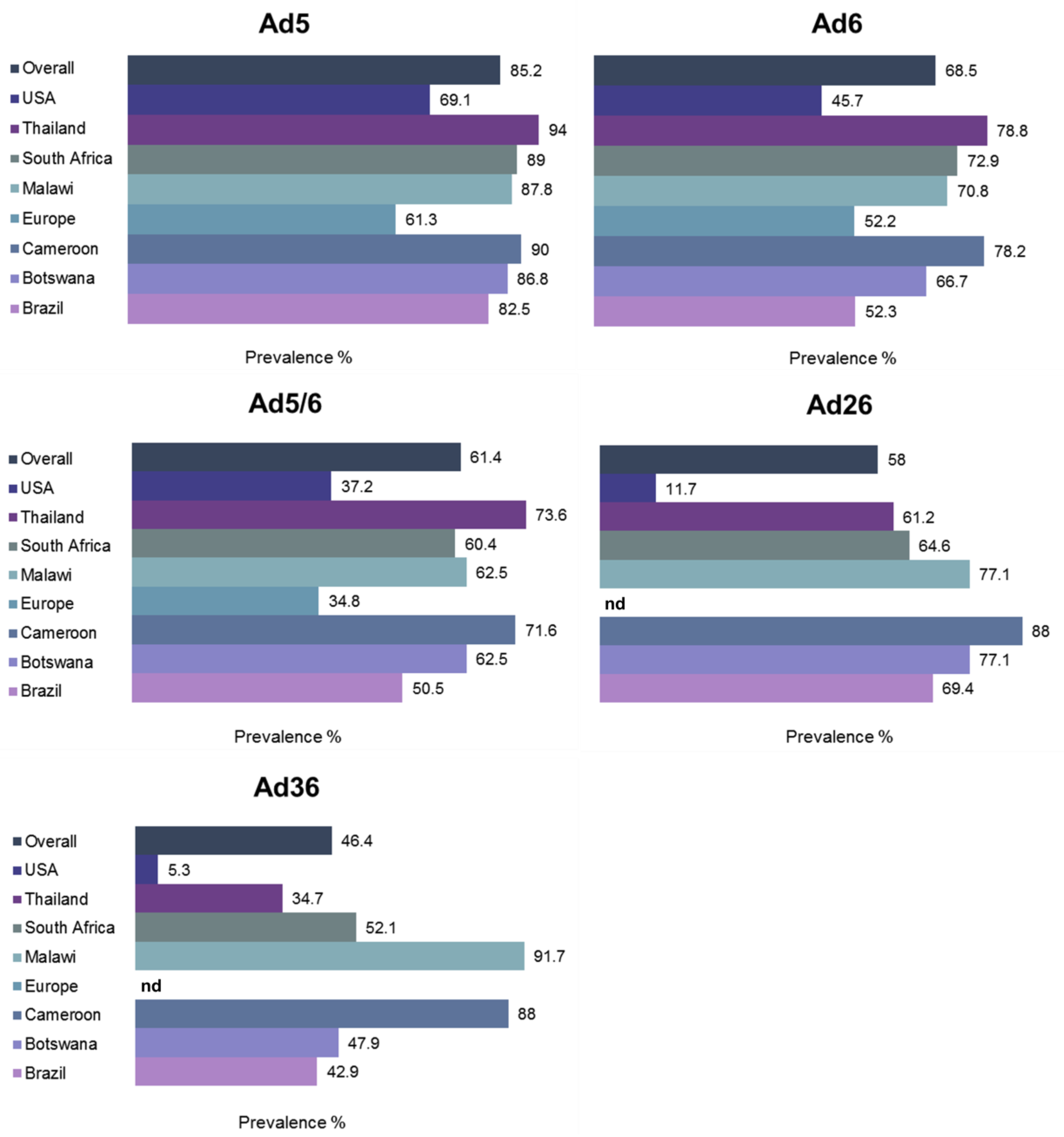
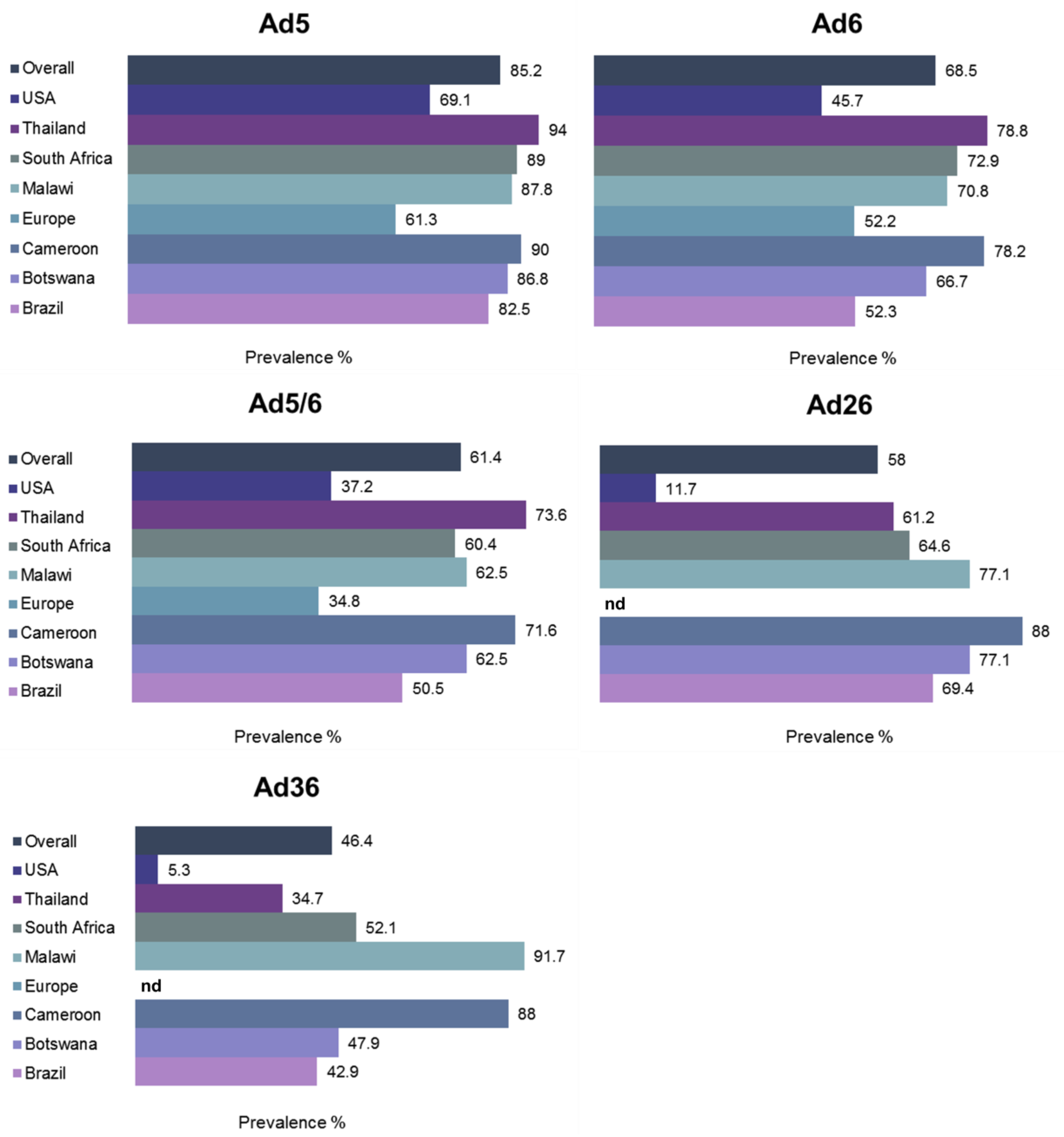
1.7. Prevalence of Pre-Existing Humoral Immunity
1.8. Vector Neutralization by Ovarian Ascites
1.9. Oncolytic Adenoviruses—the Clinical History
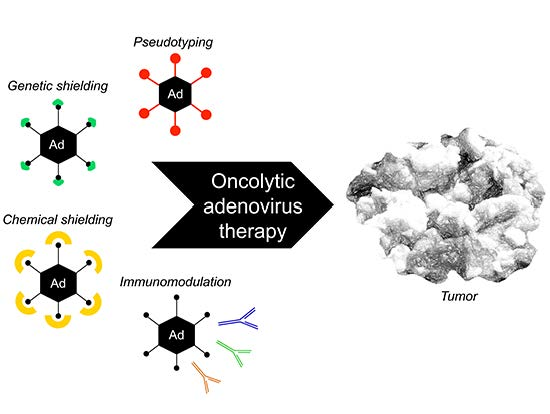
2. Therapeutic Vector Design for Host Immune Evasion
2.1. Introduction
2.2. Ads with Low Seroprevalence
2.2.1. Pseudotyped/Chimeric Vectors
2.2.2. Non-Human Vectors
2.3. Genetic Masking
2.3.1. Heterologous Peptide Incorporation within the Fiber
2.3.2. Fiber Deknobbing
2.4. Chemical Shielding
2.4.1. PEGylated Polymeric Carriers
2.4.2. Bio-Reducible (Cationic) Polymers
2.4.3. Liposomes
2.4.4. Mechanical Means of Ad Delivery into Tumors
2.4.5. Bi-Specific Adapter Molecules
3. Vector Design for Immuno-Oncolytic Therapies
3.1. Introduction
3.2. Cancer Vaccines
3.3. Immune Checkpoint Blockade
4. Perspectives
Acknowledgments
Conflicts of Interest
References
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [PubMed]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular evolution of human adenoviruses. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, S. Virus treatment questioned after gene therapy death. Nature 1999, 401, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.; Huebner, R.J.; Gilmore, L.K.; Parrott, R.H.; Ward, T.G. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc. Soc. Exp. Biol. Med. 1953, 84, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, A.; Hemminki, A. Adenoviruses for treatment of cancer. Ann. Med. 2005, 37, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Lee, J.S.; Kim, S.W.; Yun, C.O. Evolution of oncolytic adenovirus for cancer treatment. Adv. Drug Deliv. Rev. 2012, 64, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Tumor immunotherapy directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 18 November 2015).
- Abbink, P.; Lemckert, A.A.C.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; et al. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 2007, 81, 4654–4663. [Google Scholar] [CrossRef] [PubMed]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E.; et al. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Ng’ang’a, D.; Brandariz, K.L.; Abbink, P.; et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Bergelson, J.M. CAR: A virus receptor within the tight junction. Adv. Drug Deliv. Rev. 2005, 57, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.P.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.C.; Fisher, K.D.; et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting coxsackie virus-adenovirus receptor and complement receptor 1. Blood 2009, 113, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Seiradake, E.; Henaff, D.; Wodrich, H.; Billet, O.; Perreau, M.; Hippert, C.; Mennechet, F.; Schoehn, G.; Lortat-Jacob, H.; Dreja, H.; et al. The cell adhesion molecule “CAR” And sialic acid on human erythrocytes influence adenovirus in vivo biodistribution. PLoS Pathog. 2009, 5, e1000277. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.W.; Freimuth, P.; Moninger, T.O.; Ganske, I.; Zabner, J.; Welsh, M.J. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 2002, 110, 789–799. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Moller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liaw, Y.C.; Stone, D.; Kalyuzhniy, O.; Amiraslanov, I.; Tuve, S.; Verlinde, C.L.; Shayakhmetov, D.; Stehle, T.; Roffler, S.; et al. Identification of CD46 binding sites within the adenovirus serotype 35 fiber knob. J. Virol. 2007, 81, 12785–12792. [Google Scholar] [CrossRef] [PubMed]
- Kirby, I.; Davison, E.; Beavil, A.J.; Soh, C.P.C.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J. Virol. 2000, 74, 2804–2813. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Guilligay, D.; Cusack, S.; Wadell, G.; Arnberg, N. Crystal structure of species D adenovirus fiber knobs and their sialic acid binding sites. J. Virol. 2004, 78, 7727–7736. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xiang, Z.Q.; Li, Y.; Kurupati, R.K.; Jia, B.; Bian, A.; Zhou, D.M.; Hutnick, N.; Yuan, S.; Gray, C.; et al. Adenovirus-based vaccines: Comparison of vectors from three species of adenoviridae. J. Virol. 2010, 84, 10522–10532. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rhee, E.G.; Masek-Hammerman, K.; Teigler, J.E.; Abbink, P.; Barouch, D.H. Adenovirus serotype 26 utilizes CD46 as a primary cellular receptor and only transiently activates T lymphocytes following vaccination of rhesus monkeys. J. Virol. 2012, 86, 10862–10865. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.C.; Storm, R.J.; Bauer, J.; Johansson, S.M.; Lookene, A.; Angstrom, J.; Hedenstrom, M.; Eriksson, T.L.; Frangsmyr, L.; Rinaldi, S.; et al. The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nat. Med. 2011, 17, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Alba, R.; Bradshaw, A.C.; Parker, A.L.; Bhella, D.; Waddington, S.N.; Nicklin, S.A.; van Rooijen, N.; Custers, J.; Goudsmit, J.; Barouch, D.H.; et al. Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: Effect of mutagenesis on FX interactions and gene transfer. Blood 2009, 114, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Alba, R.; Parker, A.L.; Bradshaw, A.C.; McNeish, I.A.; Nicklin, S.A.; Baker, A.H. Tropism-modification strategies for targeted gene delivery using adenoviral vectors. Viruses 2010, 2, 2290–2355. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H.; et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; Parker, A.L.; Havenga, M.; Nicklin, S.A.; Buckley, S.M.K.; McVey, J.H.; Baker, A.H. Targeting of adenovirus serotype 5 (Ad5) and 5/47 pseudotyped vectors in vivo: Fundamental involvement of coagulation factors and redundancy of CAR binding by Ad5. J. Virol. 2007, 81, 9568–9571. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Baker, A.H.; Waddington, S.N.; Buckley, S.M.K.; Custers, J.; Havenga, M.J.E.; van Rooijen, N.; Goudsmit, J.; McVey, J.H.; Nicklin, S.A. Effect of neutralizing sera on factor X-mediated adenovirus serotype 5 gene transfer. J. Virol. 2009, 83, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhniy, O.; di Paolo, N.C.; Silvestry, M.; Hofherr, S.E.; Barry, M.A.; Stewart, P.L.; Shayakhmetov, D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 5483–5488. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Qiu, Q.; Tian, J.; Smith, J.S.; Conenello, G.M.; Morita, T.; Byrnes, A.P. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 2013, 19, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Khare, R.; Reddy, V.S.; Nemerow, G.R.; Barry, M.A. Identification of adenovirus serotype 5 hexon regions that interact with scavenger receptors. J. Virol. 2012, 86, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Sumida, S.M.; Truitt, D.M.; Lemckert, A.A.C.; Vogels, R.; Custers, J.H.H.V.; Addo, M.M.; Lockman, S.; Peter, T.; Peyerl, F.W.; Kishko, M.G.; et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J. Immunol. 2005, 174, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.E.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Varghese, R.; Mikyas, Y.; Stewart, P.L.; Ralston, R. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J. Virol. 2004, 78, 12320–12332. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Flatt, J.W.; di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation factor X activates innate immunity to human species C adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Irons, E.E.; Flatt, J.W.; Doronin, K.; Fox, T.L.; Acchione, M.; Stewart, P.L.; Shayakhmetov, D.M. Coagulation factor binding orientation and dimerization may influence infectivity of adenovirus-coagulation factor complexes. J. Virol. 2013, 87, 9610–9619. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Duffy, M.R.; Deng, L.; Dakin, R.S.; Uil, T.; Custers, J.; Kelly, S.M.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Manipulating adenovirus hexon hypervariable loops dictates immune neutralisation and coagulation factor X-dependent cell interaction in vitro and in vivo. PLoS Pathog. 2015, 11, e1004673. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.R.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus serotype 5 neutralizing antibodies target both hexon and fiber following vaccination and natural infection. J. Virol. 2012, 86, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.R.; Maxfield, L.F.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus serotype 5-specific neutralizing antibodies target multiple hexon hypervariable regions. J. Virol. 2012, 86, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Madisch, I.; Harste, G.; Pommer, H.; Heim, A. Phylogenetic analysis of the main neutralization and hemagglutination determinants of all human adenovirus prototypes as a basis for molecular classification and taxonomy. J. Virol. 2005, 79, 15265–15276. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Henry, L.J.; Gerard, R.D.; Deisenhofer, J. Crystal structure of the receptor-binding domain of adenovirus type 5 fiber protein at 1.7 å resolution. Structure 1994, 2, 1259–1270. [Google Scholar] [CrossRef]
- Wu, E.; Pache, L.; von Seggern, D.J.; Mullen, T.M.; Mikyas, Y.; Stewart, P.L.; Nemerow, G.R. Flexibility of the adenovirus fiber is required for efficient receptor interaction. J. Virol. 2003, 77, 7225–7235. [Google Scholar] [CrossRef] [PubMed]
- Kritz, A.B.; Nicol, C.G.; Dishart, K.L.; Nelson, R.; Holbeck, S.; von Seggern, D.J.; Work, L.M.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Adenovirus 5 fibers mutated at the putative HSPG-binding site show restricted retargeting with targeting peptides in the HI loop. Mol. Ther. 2007, 15, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.A.; Idamakanti, N.; Rollence, M.L.; Marshall-Neff, J.; Kim, J.; Mulgrew, K.; Nemerow, G.R.; Kaleko, M.; Stevenson, S.C. Adenovirus serotype 5 fiber shaft influences in vivo gene transfer in mice. Hum. Gene Ther. 2003, 14, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Bayo-Puxan, N.; Cascallo, M.; Gros, A.; Huch, M.; Fillat, C.; Alemany, R. Role of the putative heparan sulfate glycosaminoglycan-binding site of the adenovirus type 5 fiber shaft on liver detargeting and knob-mediated retargeting. J. Gen. Virol. 2006, 87, 2487–2495. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; McVey, J.H.; Doctor, J.H.; Lopez-Franco, O.; Waddington, S.N.; Havenga, M.J.; Nicklin, S.A.; Baker, A.H. Influence of coagulation factor zymogens on the infectivity of adenoviruses pseudotyped with fibers from subgroup D. J. Virol. 2007, 81, 3627–3631. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Kalyuzhniy, O.; Shayakhmetov, D.M. Fiber shaft-chimeric adenovirus vectors lacking the KKTK motif efficiently infect liver cells in vivo. J. Virol. 2007, 81, 12249–12259. [Google Scholar] [CrossRef] [PubMed]
- Corjon, S.; Gonzalez, G.; Henning, P.; Grichine, A.; Lindholm, L.; Boulanger, P.; Fender, P.; Hong, S.S. Cell entry and trafficking of human adenovirus bound to blood factor X is determined by the fiber serotype and not hexon: Heparan sulfate interaction. PLoS ONE 2011, 6, e18205. [Google Scholar] [CrossRef] [PubMed]
- Myhre, S.; Henning, P.; Granio, O.; Tylo, A.S.; Nygren, P.A.; Olofsson, S.; Boulanger, P.; Lindholm, L.; Hong, S.S. Decreased immune reactivity towards a knobless, affibody-targeted adenovirus type 5 vector. Gene Ther. 2007, 14, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Särkioja, M.; Pesonen, S.; Raki, M.; Hakkarainen, T.; Salo, J.; Ahonen, M.T.; Kanerva, A.; Hemminki, A. Changing the adenovirus fiber for retaining gene delivery efficacy in the presence of neutralizing antibodies. Gene Ther. 2008, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.; Kass-Eisler, A.; Leinwand, L.; Falck-Pedersen, E. Adenovirus type 5 and 7 capsid chimera: Fiber replacement alters receptor tropism without affecting primary immune neutralization epitopes. J. Virol. 1996, 70, 2116–2123. [Google Scholar] [PubMed]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins αvβ3and αvβ5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Neumann, R.; Chroboczek, J.; Jacrot, B. Determination of the nucleotide sequence for the penton-base gene of human adenovirus type 5. Gene 1988, 69, 153–157. [Google Scholar] [CrossRef]
- Zubieta, C.; Schoehn, G.; Chroboczek, J.; Cusack, S. The structure of the human adenovirus 2 penton. Mol. Cell 2005, 17, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.S.; Habib, N.A.; Franqueville, L.; Jensen, S.; Boulanger, P.A. Identification of adenovirus (Ad) penton base neutralizing epitopes by use of sera from patients who had received conditionally replicative Ad (Addl1520) for treatment of liver tumors. J. Virol. 2003, 77, 10366–10375. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Dong, X.; Wu, X.; Wen, B.; Ji, G.; Cheng, L.; Liu, H. Conserved fiber-penton base interaction revealed by nearly atomic resolution cryo-electron microscopy of the structure of adenovirus provides insight into receptor interaction. J. Virol. 2012, 86, 12322–12329. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Miao, E.A.; Iwakura, Y.; Murali-Krishna, K.; Aderem, A.; Flavell, R.A.; Papayannopoulou, T.; Shayakhmetov, D.M. Virus binding to a plasma membrane receptor triggers interleukin-1α-mediated proinflammatory macrophage response in vivo. Immunity 2009, 31, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Chroboczek, J.; Ruigrok, R.W.; Cusack, S. Adenovirus fiber. Curr. Top. Microbiol. Immunol. 1995, 199 Pt 1, 163–200. [Google Scholar] [PubMed]
- Shayakhmetov, D.M.; Eberly, A.M.; Li, Z.Y.; Lieber, A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005, 79, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.C.; Coughlan, L.; Miller, A.M.; Alba, R.; van Rooijen, N.; Nicklin, S.A.; Baker, A.H. Biodistribution and inflammatory profiles of novel penton and hexon double-mutant serotype 5 adenoviruses. J. Control. Release 2012, 164, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Appledorn, D.M.; Amalfitano, A. Adenovirus vector induced innate immune responses: Impact upon efficacy and toxicity in gene therapy and vaccine applications. Virus Res. 2008, 132, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate immunity to adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Cichon, G.; Boeckh-Herwig, S.; Schmidt, H.H.; Wehnes, E.; Müller, T.; Pring-Akerblom, P.; Burger, R. Complement activation by recombinant adenoviruses. Gene Ther. 2001, 8, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Appledorn, D.M.; McBride, A.; Seregin, S.; Scott, J.M.; Schuldt, N.; Kiang, A.; Godbehere, S.; Amalfitano, A. Complex interactions with several arms of the complement system dictate innate and humoral immunity to adenoviral vectors. Gene Ther. 2008, 15, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbein, A.F.; Fehr, T.; Lutz, C.; Suter, M.; Brombacher, F.; Hengartner, H.; Zinkernagel, R.M. Control of early viral and bacterial distribution and disease by natural antibodies. Science 1999, 286, 2156–2159. [Google Scholar] [CrossRef] [PubMed]
- Othman, M.; Labelle, A.; Mazzetti, I.; Elbatarny, H.S.; Lillicrap, D. Adenovirus-induced thrombocytopenia: The role of von Willebrand factor and p-selectin in mediating accelerated platelet clearance. Blood 2007, 109, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Idamakanti, N.; Kylefjord, H.; Rollence, M.; King, L.; Kaloss, M.; Kaleko, M.; Stevenson, S.C. In vivo hepatic adenoviral gene delivery occurs independently of the coxsackievirus-adenovirus receptor. Mol. Ther. 2002, 5, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef] [PubMed]
- Juillard, V.; Villefroy, P.; Godfrin, D.; Pavirani, A.; Venet, A.; Guillet, J.G. Long-term humoral and cellular immunity induced by a single immunization with replication-defective adenovirus recombinant vector. Eur. J. Immunol. 1995, 25, 3467–3473. [Google Scholar] [CrossRef] [PubMed]
- Jooss, K.; Chirmule, N. Immunity to adenovirus and adeno-associated viral vectors: Implications for gene therapy. Gene Ther. 2003, 10, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Tomasec, P.; Wang, E.C.Y.; Groh, V.; Spies, T.; McSharry, B.P.; Aicheler, R.J.; Stanton, R.J.; Wilkinson, G.W.G. Adenovirus vector delivery stimulates natural killer cell recognition. J. Gen. Virol. 2007, 88, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Sumida, S.M.; Truitt, D.M.; Kishko, M.G.; Arthur, J.C.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Koudstaal, W.; Pau, M.G.; Kostense, S.; et al. Neutralizing antibodies and CD8+ T lymphocytes both contribute to immunity to adenovirus serotype 5 vaccine vectors. J. Virol. 2004, 78, 2666–2673. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Eisenlohr, L.; Flomenberg, N.; Hsu, S.; Flomenberg, P. The adenovirus capsid protein hexon contains a highly conserved human CD4+ T-cell epitope. Hum. Gene Ther. 2002, 13, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Onion, D.; Crompton, L.J.; Milligan, D.W.; Moss, P.A.H.; Lee, S.P.; Mautner, V. The CD4+ T-cell response to adenovirus is focused against conserved residues within the hexon protein. J. Gen. Virol. 2007, 88, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Ariyawansa, J.P.; Tobin, J.O. Fluorescent antibody responses to adenoviruses in humans. J. Clin. Pathol. 1976, 29, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Crawford-Miksza, L.; Schnurr, D.P. Seroepidemiology of new AIDS-associated adenoviruses among the San Francisco men’s health study. J. Med. Virol. 1996, 50, 230–236. [Google Scholar] [CrossRef]
- Harvey, B.G.; Hackett, N.R.; El-Sawy, T.; Rosengart, T.K.; Hirschowitz, E.A.; Lieberman, M.D.; Lesser, M.L.; Crystal, R.G. Variability of human systemic humoral immune responses to adenovirus gene transfer vectors administered to different organs. J. Virol. 1999, 73, 6729–6742. [Google Scholar] [PubMed]
- Sharma, A.; Tandon, M.; Ahi, Y.S.; Bangari, D.S.; Vemulapalli, R.; Mittal, S.K. Evaluation of cross-reactive cell-mediated immune responses among human, bovine and porcine adenoviruses. Gene Ther. 2010, 17, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, C.E.G.; Svensson, U.K.; Everitt, E. Interaction between HeLa cells and adenovirus type 2 virions neutralized by different antisera. J. Virol. 1985, 56, 896–903. [Google Scholar] [PubMed]
- Wohlfart, C. Neutralization of adenoviruses: Kinetics, stoichiometry, and mechanisms. J. Virol. 1988, 62, 2321–2328. [Google Scholar] [PubMed]
- Gahéry-Ségard, H.; Farace, F.; Godfrin, D.; Gaston, J.; Lengagne, R.; Tursz, T.; Boulanger, P.; Guillet, J.G. Immune response to recombinant capsid proteins of adenovirus in humans: Antifiber and anti-penton base antibodies have a synergistic effect on neutralizing activity. J. Virol. 1998, 72, 2388–2397. [Google Scholar] [PubMed]
- Cheng, C.; Gall, J.G.D.; Nason, M.; King, C.R.; Koup, R.A.; Roederer, M.; McElrath, M.J.; Morgan, C.A.; Churchyard, G.; Baden, L.R.; et al. Differential specificity and immunogenicity of adenovirus type 5 neutralizing antibodies elicited by natural infection or immunization. J. Virol. 2010, 84, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the STEP study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef]
- Überla, K. HIV vaccine development in the aftermath of the STEP study: Re-focus on occult HIV infection? PLoS Pathog. 2008, 4, e1000114. [Google Scholar] [CrossRef] [PubMed]
- Nwanegbo, E.; Vardas, E.; Gao, W.; Whittle, H.; Sun, H.; Rowe, D.; Robbins, P.D.; Gambotto, A. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of the Gambia, South Africa, and The United States. Clin. Diagn. Lab. Immunol. 2004, 11, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Thorner, A.R.; Vogels, R.; Kaspers, J.; Weverling, G.J.; Holterman, L.; Lemckert, A.A.C.; Dilraj, A.; McNally, L.M.; Jeena, P.M.; Jepsen, S.; et al. Age dependence of adenovirus-specific neutralizing antibody titers in individuals from sub-Saharan Africa. J. Clin. Microbiol. 2006, 44, 3781–3783. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Heath and Care Excellence. Ovarian Cancer: The Recognition and Initial Management of Ovarian Cancer. Available online: http://www.nice.org.uk/guidance/CG122/chapter/introduction (accessed on 18 November 2015).
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Coping with Cancer—Treating Fluid in the Abdomen. Available online: http://www.cancerresearchuk.org/about-cancer/coping-with-cancer/coping-physically/fluid-in-the-abdomen-ascites/treating-fluid-in-abdomen#drain (accessed on 18 November 2015).
- Hemminki, A.; Wang, M.; Desmond, R.A.; Strong, T.V.; Alvarez, R.D.; Curiel, D.T. Serum and ascites neutralizing antibodies in ovarian cancer patients treated with intraperitoneal adenoviral gene therapy. Hum. Gene Ther. 2002, 13, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Stallwood, Y.; Fisher, K.D.; Gallimore, P.H.; Mautner, V. Neutralisation of adenovirus infectivity by ascitic fluid from ovarian cancer patients. Gene Ther. 2000, 7, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.L.; Hui, L.; Gomez-Navarro, J.; Dmitriev, I.; Krasnykh, V.; Richter, C.A.; Shaw, D.R.; Alvarez, R.D.; Curiel, D.T.; Strong, T.V. Using a tropism-modified adenoviral vector to circumvent inhibitory factors in ascites fluid. Hum. Gene Ther. 2000, 11, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, T.G.; Thériault, B.L.; Campbell, E.J.; Nachtigal, M.W. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat. Protoc. 2007, 1, 2643–2649. [Google Scholar] [CrossRef] [PubMed]
- Thériault, B.L.; Portelance, L.; Mes-Masson, A.M.; Nachtigal, M.W. Establishment of primary cultures from ovarian tumor tissue and ascites fluid. Methods Mol. Biol. 2013, 1049, 323–336. [Google Scholar] [PubMed]
- O’Donnell, R.L.; McCormick, A.; Mukhopadhyay, A.; Woodhouse, L.C.; Moat, M.; Grundy, A.; Dixon, M.; Kaufman, A.; Soohoo, S.; Elattar, A.; et al. The use of ovarian cancer cells from patients undergoing surgery to generate primary cultures capable of undergoing functional analysis. PLoS ONE 2014, 9, e90604. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmitz, G.J.; Barker, S.D.; Hemminki, A. Adenoviral gene therapy for cancer: From vectors to targeted and replication competent agents. Int. J. Oncol. 2002, 21, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Kirn, D.; Eckhardt, S.G.; Rodriguez, G.I.; Soutar, D.S.; Otto, R.; Robertson, A.G.; Park, O.; Gulley, M.L.; Heise, C.; et al. A phase I study of ONYX-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin. Cancer Res. 2000, 6, 798–806. [Google Scholar] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Bergh, J.; Torbjjrn, N.; Sjogren, S.; Lindgren, A.; Holmberg, L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995, 1, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in Tp53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1674. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.Y.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Cascallo, M.; Alonso, M.M.; Rojas, J.J.; Perez-Gimenez, A.; Fueyo, J.; Alemany, R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRb pathway. Mol. Ther. 2007, 15, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Nokisalmi, P.; Pesonen, S.; Escutenaire, S.; Särkioja, M.; Raki, M.; Cerullo, V.; Laasonen, L.; Alemany, R.; Rojas, J.; Cascallo, M.; et al. Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin. Cancer Res. 2010, 16, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Post, D.E.; van Meir, E.G. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene 2003, 22, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; van Ginkel, J.W.; Au, K.Y.; Alemany, R.; Meulenberg, J.J.; van Beusechem, V.W. ORCA-010, a novel potency-enhanced oncolytic adenovirus, exerts strong antitumor activity in preclinical models. Hum. Gene Ther. 2014, 25, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Martínez-Quintanilla, J.; Puig, C.; Guedan, S.; Molleví, D.G.; Alemany, R.; Cascallo, M. Bioselection of a gain of function mutation that enhances adenovirus 5 release and improves its antitumoral potency. Cancer Res. 2008, 68, 8928–8937. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.; Ranki, T.; Joensuu, T.; Jager, E.; Karbach, J.; Wahle, C.; Partanen, K.; Kairemo, K.; Alanko, T.; Turkki, R.; et al. Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8 T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef] [PubMed]
- Branton, P.E.; Roopchand, D.E. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene 2001, 20, 7855–7865. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Seymour, L.; Fisher, K. Activity of a group B oncolytic adenovirus (ColoAd1) in whole human blood. Gene Ther. 2014, 21, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Xin, Y.; Zheng, J.N.; Liu, Y.Q. Combining conditionally replicating adenovirus-mediated gene therapy with chemotherapy: A novel antitumor approach. Int. J. Cancer 2011, 129, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.L.; Post, D.E.; Khuri, F.R.; van Meir, E.G. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin. Cancer Res. 2004, 10, 5299–5312. [Google Scholar] [CrossRef] [PubMed]
- Jennings, V.A.; Ilett, E.J.; Scott, K.J.; West, E.J.; Vile, R.; Pandha, H.; Harrington, K.; Young, A.; Hall, G.D.; Coffey, M.; et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int. J. Cancer 2014, 134, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pong, R.C.; Bergelson, J.M.; Hall, M.C.; Sagalowsky, A.I.; Tseng, C.P.; Wang, Z.; Hsieh, J.T. Loss of adenoviral receptor expression in human bladder cancer cells: A potential impact on the efficacy of gene therapy. Cancer Res. 1999, 59, 325–330. [Google Scholar] [PubMed]
- Wunder, T.; Schumacher, U.; Friedrich, R.E. Coxsackie adenovirus receptor expression in carcinomas of the head and neck. Anticancer Res. 2012, 32, 1057–1062. [Google Scholar] [PubMed]
- Kaufmann, J.K.; Nettelbeck, D.M. Virus chimeras for gene therapy, vaccination, and oncolysis: Adenoviruses and beyond. Trends Mol. Med. 2012, 18, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Ophorst, O.J.A.E.; Kostense, S.; Goudsmit, J.; de Swart, R.L.; Verhaagh, S.; Zakhartchouk, A.; van Meijer, M.; Sprangers, M.; van Amerongen, G.; Yüksel, S.; et al. An adenoviral type 5 vector carrying a type 35 fiber as a vaccine vehicle: DC targeting, cross neutralization, and immunogenicity. Vaccine 2004, 22, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Provine, N.M.; Ra, J.; Borducchi, E.N.; McNally, A.; Simmons, N.L.; Iampietro, M.J.; Barouch, D.H. Alternative serotype adenovirus vaccine vectors elicit memory T cells with enhanced anamnestic capacity compared to Ad5 vectors. J. Virol. 2013, 87, 1373–1384. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uchino, J.; Curiel, D.T.; Ugai, H. Species D human adenovirus type 9 exhibits better virus-spread ability for antitumor efficacy among alternative serotypes. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Teigler, J.E.; Iampietro, M.J.; Barouch, D.H. Vaccination with adenovirus serotypes 35, 26, and 48 elicits higher levels of innate cytokine responses than adenovirus serotype 5 in rhesus monkeys. J. Virol. 2012, 86, 9590–9598. [Google Scholar] [CrossRef] [PubMed]
- Camacho, Z.T.; Turner, M.A.; Barry, M.A.; Weaver, E.A. CD46-mediated transduction of a species D adenovirus vaccine improves mucosal vaccine efficacy. Hum. Gene Ther. 2014, 25, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Adams, W.C.; Bond, E.; Havenga, M.J.E.; Holterman, L.; Goudsmit, J.; Hedestam, G.B.K.; Koup, R.A.; Loré, K. Adenovirus serotype 5 infects human dendritic cells via a coxsackievirus-adenovirus receptor-independent receptor pathway mediated by lactoferrin and DC-SIGN. J. Gen. Virol. 2009, 90, 1600–1610. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Senac, J.S.; Weaver, E.A.; May, S.M.; Jelinek, D.F.; Greipp, P.; Witzig, T.; Barry, M.A. Species D adenoviruses as oncolytics against B-cell cancers. Clin. Cancer Res. 2011, 17, 6712–6722. [Google Scholar] [CrossRef] [PubMed]
- Mastrangeli, A.; Harvey, B.G.; Yao, J.; Wolff, G.; Kovesdi, I.; Crystal, R.G.; Falck-Pedersen, E. “Sero-switch” Adenovirus-mediated in vivo gene transfer: Circumvention of anti-adenovirus humoral immune defenses against repeat adenovirus vector administration by changing the adenovirus serotype. Hum. Gene Ther. 1996, 7, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.A.; Song, W.R.; Carpenter, H.; Wickham, T.J.; Kovesdi, I.; Harvey, B.G.; Magovern, C.J.; Isom, O.W.; Rosengart, T.; Falck-Pedersen, E.; et al. Circumvention of anti-adenovirus neutralizing immunity by administration of an adenoviral vector of an alternate serotype. Hum. Gene Ther. 1997, 8, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Bradshaw, A.C.; Parker, A.L.; Robinson, H.; White, K.; Custers, J.; Goudsmit, J.; van Roijen, N.; Barouch, D.H.; Nicklin, S.A.; et al. Ad5:Ad48 hexon hypervariable region substitutions lead to toxicity and increased inflammatory responses following intravenous delivery. Mol. Ther. 2012, 20, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Teigler, J.E.; Penaloza-MacMaster, P.; Obeng, R.; Provine, N.M.; Larocca, R.A.; Borducchi, E.N.; Barouch, D.H. Hexon hypervariable region-modified adenovirus type 5 (Ad5) vectors display reduced hepatotoxicity but induce T lymphocyte phenotypes similar to Ad5 vectors. Clin. Vaccine Immunol. 2014, 21, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Nociari, M.; Philpott, N.; Falck-Pedersen, E. Influence of fiber detargeting on adenovirus-mediated innate and adaptive immune activation. J. Virol. 2005, 79, 11627–11637. [Google Scholar] [CrossRef] [PubMed]
- Rogee, S.; Grellier, E.; Bernard, C.; Jouy, N.; Loyens, A.; Beauvillain, J.C.; Fender, P.; Corjon, S.; Hong, S.S.; Boulanger, P.; et al. Influence of chimeric human-bovine fibers on adenoviral uptake by liver cells and the antiviral immune response. Gene Ther. 2010, 17, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Denby, L.; Work, L.M.; Graham, D.; Hsu, C.; von Seggern, D.J.; Nicklin, S.A.; Baker, A.H. Adenoviral serotype 5 vectors pseudotyped with fibers from subgroup D show modified tropism in vitro and in vivo. Hum. Gene Ther. 2004, 15, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- White, K.M.; Alba, R.; Parker, A.L.; Wright, A.F.; Bradshaw, A.C.; Delles, C.; McDonald, R.A.; Baker, A.H. Assessment of a novel, capsid-modified adenovirus with an improved vascular gene transfer profile. J. Cardiothorac. Surg. 2013, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Alba, R.; Bradshaw, A.C.; Coughlan, L.; Denby, L.; McDonald, R.A.; Waddington, S.N.; Buckley, S.M.K.; Greig, J.A.; Parker, A.L.; Miller, A.M.; et al. Biodistribution and retargeting of FX-binding ablated adenovirus serotype 5 vectors. Blood 2010, 116, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; White, K.M.; Lavery, C.A.; Custers, J.; Waddington, S.N.; Baker, A.H. Pseudotyping the adenovirus serotype 5 capsid with both the fibre and penton of serotype 35 enhances vascular smooth muscle cell transduction. Gene Ther. 2013, 20, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gordo, E.; Podgorski, II; Downes, N.; Alemany, R. Circumventing antivector immunity: Potential use of nonhuman adenoviral vectors. Hum. Gene Ther. 2014, 25, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Junyent, F.; Kremer, E.J. CAV-2—Why a canine virus is a neurobiologist’s best friend. Curr. Opin. Pharmacol. 2015, 24, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Löser, P.; Cichon, G.; Arnold, W.; Both, G.W.; Strauss, M. Ovine adenovirus vectors overcome preexisting humoral immunity against human adenoviruses in vivo. J. Virol. 1999, 73, 6930–6936. [Google Scholar] [PubMed]
- Bangari, D.S.; Mittal, S.K. Porcine adenoviral vectors evade preexisting humoral immunity to adenoviruses and efficiently infect both human and murine cells in culture. Virus Res. 2004, 105, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, S.; Hays, J.; Hogenesch, H.; Mittal, S.K. Circumvention of vector-specific neutralizing antibody response by alternating use of human and non-human adenoviruses: Implications in gene therapy. Virology 2000, 272, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, J.N.; Kremer, E.J.; Hemminki, A.; Siegal, G.P.; Douglas, J.T.; Curiel, D.T. An adenovirus vector with a chimeric fiber derived from canine adenovirus type 2 displays novel tropism. Virology 2004, 324, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Reetz, J.; Herchenroder, O.; Putzer, B.M. Peptide-based technologies to alter adenoviral vector tropism: Ways and means for systemic treatment of cancer. Viruses 2014, 6, 1540–1563. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Kerttula, H.; Legut, M.; Davies, J.; Jones, R.; Hudson, E.; Hanna, L.; Stanton, R.J.; Chester, J.D.; Parker, A.L. Incorporation of peptides targeting EGFR and FGFR1 into the adenoviral fiber knob domain and their evaluation as targeted cancer therapies. Hum. Gene Ther. 2015, 26, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.K.; Hong, S.S.; Boulanger, P.; Lindholm, L. Genetic retargeting of adenovirus: Novel strategy employing “deknobbing” of the fiber. J. Virol. 2001, 75, 7280–7289. [Google Scholar] [CrossRef] [PubMed]
- Belousova, N.; Mikheeva, G.; Gelovani, J.; Krasnykh, V. Modification of adenovirus capsid with a designed protein ligand yields a gene vector targeted to a major molecular marker of cancer. J. Virol. 2008, 82, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, Z.; de Vrij, J.; Magnusson, M.; Debets, R.; Willemsen, R. An oncolytic adenovirus redirected with a tumor-specific T-cell receptor. Cancer Res. 2007, 67, 11309–11316. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targrts 2007, 7, 141–148. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Cunningham, C.; Buchanan, A.; Blackburn, A.; Edelman, G.; Maples, P.; Netto, G.; Tong, A.; Randlev, B.; Olson, S.; et al. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: Safety, feasibility and biological activity. Gene Ther. 2001, 8, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Warren, R.; Kirn, D. Intravascular adenoviral agents in cancer patients: Lessons from clinical trials. Cancer Gene Ther. 2002, 9, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Carducci, M.A.; Burke, J.M.; Rodriguez, R.; Fong, L.; van Ummersen, L.; Yu, D.C.; Aimi, J.; Ando, D.; Working, P.; et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol. Ther. 2006, 14, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Sachs, M.D.; Rauen, K.A.; Ramamurthy, M.; Dodson, J.L.; de Marzo, A.M.; Putzi, M.J.; Schoenberg, M.P.; Rodriguez, R. Integrin α v and coxsackie adenovirus receptor expression in clinical bladder cancer. Urology 2002, 60, 531–536. [Google Scholar] [CrossRef]
- You, Z.; Fischer, D.C.; Tong, X.; Hasenburg, A.; Aguilar-Cordova, E.; Kieback, D.G. Coxsackievirus-adenovirus receptor expression in ovarian cancer cell lines is associated with increased adenovirus transduction efficiency and transgene expression. Cancer Gene Ther. 2001, 8, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Zinn, K.R.; Barnett, B.G.; Sumerel, L.A.; Krasnykh, V.; Curiel, D.T.; Douglas, J.T. The therapeutic efficacy of adenoviral vectors for cancer gene therapy is limited by a low level of primary adenovirus receptors on tumour cells. Eur. J. Cancer 2002, 38, 1917–1926. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, S.H.; Cho, Y.S.; Choi, J.J.; Kim, Y.H.; Lee, J.H. Enhancement of the adenoviral sensitivity of human ovarian cancer cells by transient expression of coxsackievirus and adenovirus receptor (CAR). Gynecol. Oncol. 2002, 85, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, K.; Sundaresan, A.; Cheow, W.S. Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: A review. Eur. J. Pharm. Biopharm. 2013, 85, 427–443. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, C.R.; Lachapelle, A.; Delgado, C.; Parkes, V.; Wadsworth, S.C.; Smith, A.E.; Francis, G.E. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum. Gene Ther. 1999, 10, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Chillón, M.; Lee, J.H.; Fasbender, A.; Welsh, M.J. Adenovirus complexed with polyethylene glycol and cationic lipid is shielded from neutralizing antibodies in vitro. Gene Ther. 1998, 5, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Eto, Y.; Gao, J.Q.; Sekiguchi, F.; Kurachi, S.; Katayama, K.; Mizuguchi, H.; Hayakawa, T.; Tsutsumi, Y.; Mayumi, T.; Nakagawa, S. Neutralizing antibody evasion ability of adenovirus vector induced by the bioconjugation of methoxypolyethylene glycol succinimidyl propionate (mPEG-SPA). Biol. Pharm. Bull. 2004, 27, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Eto, Y.; Yoshioka, Y.; Ishida, T.; Yao, X.; Morishige, T.; Narimatsu, S.; Mizuguchi, H.; Mukai, Y.; Okada, N.; Kiwad, H.; et al. Optimized PEGylated adenovirus vector reduces the anti-vector humoral immune response against adenovirus and induces a therapeutic effect against metastatic lung cancer. Biol. Pharm. Bull. 2010, 33, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Sakurai, F.; Katayama, K.; Yamaguchi, T.; Okamoto, S.; Takahira, K.; Tachibana, M.; Nakagawa, S.; Mizuguchi, H. A hexon-specific pegylated adenovirus vector utilizing blood coagulation factor X. Biomaterials 2012, 33, 3743–3755. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Kochanek, S. Modification of adenovirus gene transfer vectors with synthetic polymers: A scientific review and technical guide. Mol. Ther. 2008, 16, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Kouyama, E.; Katayama, K.; Sakurai, F.; Yamaguchi, T.; Kurachi, S.; Kawabata, K.; Nakagawa, S.; Mizuguchi, H. Hexon-specific PEGylated adenovirus vectors utilizing avidin-biotin interaction. Biomaterials 2011, 32, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Prill, J.M.; Espenlaub, S.; Samen, U.; Engler, T.; Schmidt, E.; Vetrini, F.; Rosewell, A.; Grove, N.; Palmer, D.; Ng, P.; et al. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Mol. Ther. 2011, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Shashkova, E.V.; May, S.M.; Hofherr, S.E.; Barry, M.A. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum. Gene Ther. 2009, 20, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, S.W. Bioreducible polymers for therapeutic gene delivery. J. Control. Release 2014, 190, 424–439. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.H.; Kim, T.I.; Yockman, J.W.; Kim, S.W.; Yun, C.O. The effect of surface modification of adenovirus with an arginine-grafted bioreducible polymer on transduction efficiency and immunogenicity in cancer gene therapy. Biomaterials 2010, 31, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.J.; Kasala, D.; Choi, J.W.; Lee, S.H.; Hwang, J.K.; Kim, S.W.; Yun, C.O. Safety profiles and antitumor efficacy of oncolytic adenovirus coated with bioreducible polymer in the treatment of a CAR-negative tumor model. Biomacromolecules 2015, 16, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, J.; Bui, Q.N.; Li, Y.; Yun, C.O.; Lee, D.S.; Kim, S.W. Tuning surface charge and PEGylation of biocompatible polymers for efficient delivery of nucleic acid or adenoviral vector. Bioconjug. Chem. 2015, 26, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Prill, J.M.; Šubr, V.; Pasquarelli, N.; Engler, T.; Hoffmeister, A.; Kochanek, S.; Ulbrich, K.; Kreppel, F. Traceless bioresponsive shielding of adenovirus hexon with HPMA copolymers maintains transduction capacity in vitro and in vivo. PLoS ONE 2014, 9, e82716. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.D.; Stallwood, Y.; Green, N.K.; Ulbrich, K.; Mautner, V.; Seymour, L.W. Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 2001, 8, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Fisher, K.D.; Oupicky, D.; Read, M.L.; Nicklin, S.A.; Baker, A.H.; Seymour, L.W. Enhanced gene transfer activity of peptide-targeted gene-delivery vectors. J. Drug Target. 2005, 13, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nam, H.Y.; Kim, T.I.; Kim, P.H.; Ryu, J.; Yun, C.O.; Kim, S.W. Active targeting of RGD-conjugated bioreducible polymer for delivery of oncolytic adenovirus expressing shRNA against IL-8 mRNA. Biomaterials 2011, 32, 5158–5166. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, S.J.; Lim, S.J. Formulation and in vitro and in vivo evaluation of a cationic emulsion as a vehicle for improving adenoviral gene transfer. Int. J. Pharm. 2014, 475, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Coussios, C.C.; Seymour, L.; Carlisle, R. Ultrasound-enhanced drug delivery for cancer. Expert Opin. Drug Deliv. 2012, 9, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, R.; Choi, J.; Bazan-Peregrino, M.; Laga, R.; Subr, V.; Kostka, L.; Ulbrich, K.; Coussios, C.C.; Seymour, L.W. Enhanced tumor uptake and penetration of virotherapy using polymer stealthing and focused ultrasound. J. Natl. Cancer Inst. 2013, 105, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Carlisle, R.; Laga, R.; Myers, R.; Graham, S.; Cawood, R.; Ulbrich, K.; Seymour, L.; Coussios, C.C. Increasing the density of nanomedicines improves their ultrasound-mediated delivery to tumours. J. Control. Release 2015, 210, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.T.; Miller, C.R.; Kim, M.; Dmitriev, I.; Mikheeva, G.; Krasnykh, V.; Curiel, D.T. A system for the propagation of adenoviral vectors with genetically modified receptor specificities. Nat. Biotechnol. 1999, 17, 470–475. [Google Scholar] [PubMed]
- Rancourt, C.; Robertson, M.W., III; Wang, M.; Goldman, C.K.; Kelly, J.F.; Alvarez, R.D.; Siegal, G.P.; Curiel, D.T. Endothelial cell vehicles for delivery of cytotoxic genes as a gene therapy approach for carcinoma of the ovary. Clin. Cancer Res. 1998, 4, 265–270. [Google Scholar] [PubMed]
- Dmitriev, I.; Kashentseva, E.; Rogers, B.E.; Krasnykh, V.; Curiel, D.T. Ectodomain of coxsackievirus and adenovirus receptor genetically fused to epidermal growth factor mediates adenovirus targeting to epidermal growth factor receptor-positive cells. J. Virol. 2000, 74, 6875–6884. [Google Scholar] [CrossRef] [PubMed]
- Van Beusechem, V.W.; Mastenbroek, D.C.J.; van den Doel, P.B.; Lamfers, M.L.M.; Grill, J.; Würdinger, T.; Haisma, H.J.; Pinedo, H.M.; Gerritsen, W.R. Conditionally replicative adenovirus expressing a targeting adapter molecule exhibits enhanced oncolytic potency on CAR-deficient tumors. Gene Ther. 2003, 10, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; May, S.M.; Barry, M.A. Targeting adenoviruses with factor X-single-chain antibody fusion proteins. Hum. Gene Ther. 2010, 21, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Nettelbeck, D.M.; Miller, D.W.; Jerome, V.; Zuzarte, M.; Watkins, S.J.; Hawkins, R.E.; Muller, R.; Kontermann, R.E. Targeting of adenovirus to endothelial cells by a bispecific single-chain diabody directed against the adenovirus fiber knob domain and human endoglin (CD105). Mol. Ther. 2001, 3, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Graat, H.C.A.; Schagen, F.H.E.; Mastenbroek, D.C.J.; Rots, M.G.; Haisma, H.J.; Groothuis, G.M.M.; Schaap, G.R.; Bras, J.; Kaspers, G.J.L.; et al. A conditionally replicating adenovirus with strict selectivity in killing cells expressing epidermal growth factor receptor. Virology 2007, 361, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Dreier, B.; Mikheeva, G.; Belousova, N.; Parizek, P.; Boczek, E.; Jelesarov, I.; Forrer, P.; Plückthun, A.; Krasnykh, V. Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting. J. Mol. Biol. 2011, 405, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Dreier, B.; Honegger, A.; Hess, C.; Nagy-Davidescu, G.; Mittl, P.R.E.; Grütter, M.G.; Belousova, N.; Mikheeva, G.; Krasnykh, V.; Plückthun, A. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric darpin adapters. Proc. Natl. Acad. Sci. USA 2013, 110, E869–E877. [Google Scholar] [CrossRef] [PubMed]
- Harvey, T.J.; Burdon, D.; Steele, L.; Ingram, N.; Hall, G.D.; Selby, P.J.; Vile, R.G.; Cooper, P.A.; Shnyder, S.D.; Chester, J.D. Retargeted adenoviral cancer gene therapy for tumour cells overexpressing epidermal growth factor receptor or urokinase-type plasminogen activator receptor. Gene Ther. 2010, 17, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Zarour, H.; DeLeo, A.; Finn, O.; Storkus, W. Holland-Frei cancer medicine. In Categories of Tumor Antigens, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Kuroki, M.; Shirasu, N. Novel treatment strategies for cancer and their tumor-targeting approaches using antibodies against tumor-associated antigens. Anticancer Res. 2014, 34, 4481–4488. [Google Scholar] [PubMed]
- Zhang, L.; Tang, Y.; Akbulut, H.; Zelterman, D.; Linton, P.J.; Deisseroth, A.B. An adenoviral vector cancer vaccine that delivers a tumor-associated antigen/CD40-ligand fusion protein to dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15101–15106. [Google Scholar] [CrossRef] [PubMed]
- Majhen, D.; Calderon, H.; Chandra, N.; Fajardo, C.A.; Rajan, A.; Alemany, R.; Custers, J. Adenovirus-based vaccines for fighting infectious diseases and cancer: Progress in the field. Hum. Gene Ther. 2014, 25, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Gürlevik, E.; Ureche, C.I.; Schumacher, A.; Kühnel, F. Oncolytic viruses as anticancer vaccines. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Park, B.H.; Hwang, T.; Liu, T.C.; Sze, D.Y.; Kim, J.S.; Kwon, H.C.; Oh, S.Y.; Han, S.Y.; Yoon, J.H.; Hong, S.H.; et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: A phase I trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Bines, S.D. Optim trial: A phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010, 6, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Amgen announces oncolytic virus shrinks tumors. Nat. Biotech. 2013, 31, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Collichio, F.A.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.; Spitler, L.; Puzanov, I.; Agarwala, S.; Milhem, M.; et al. Final planned overall survival (OS) from Optim, a randomized phase III trial of talimogene laherparepvec (T-VEC) versus GM-CSF for the treatment of unresected stage IIIB/C/IV melanoma (nct00769704). J. Immunother. Cancer 2014, 2, P263–P263. [Google Scholar] [CrossRef]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M. Translating tumor antigens into cancer vaccines. Clin. Vaccine Immunol. 2011, 18, 23–34. [Google Scholar] [PubMed]
- Nguyen, A.; Ho, L.; Wan, Y. Chemotherapy and oncolytic virotherapy: Advanced tactics in the war against cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunelliere, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [PubMed]
- Wilkinson-Ryan, I.; Kim, J.; Kim, S.; Ak, F.; Dodson, L.; Colonna, M.; Powell, M.; Mutch, D.; Spitzer, D.; Hansen, T.; et al. Incorporation of porcine adenovirus 4 fiber protein enhances infectivity of adenovirus vector on dendritic cells: Implications for immune-mediated cancer therapy. PLoS ONE 2015, 10, e0125851. [Google Scholar] [PubMed]
- Xie, J.; Guo, X.; Liu, F.; Luo, J.; Duan, F.; Tao, X. In vitro antitumor immune response induced by dendritic cells transduced with human livin alpha recombinant adenovirus. Cell Immunol. 2015, 297, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Bauzon, M.; Hermiston, T. Armed therapeutic viruses—A disruptive therapy on the horizon of cancer immunotherapy. Front. Immunol. 2014, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Wolchok, J.D. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.J.; Sullivan, T.J.; Allison, J.P. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J. Immunol. 2006, 177, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Mohammed, S.; Al-Omair, A.; Qattan, A.; Lehe, C.; Al-Qudaihi, G.; Elkum, N.; Alshabanah, M.; Bin Amer, S.; Tulbah, A.; et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: Correlation with important high-risk prognostic factors. Neoplasia 2006, 8, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Loskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uusi-Kerttula, H.; Hulin-Curtis, S.; Davies, J.; Parker, A.L. Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications. Viruses 2015, 7, 6009-6042. https://doi.org/10.3390/v7112923
Uusi-Kerttula H, Hulin-Curtis S, Davies J, Parker AL. Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications. Viruses. 2015; 7(11):6009-6042. https://doi.org/10.3390/v7112923
Chicago/Turabian StyleUusi-Kerttula, Hanni, Sarah Hulin-Curtis, James Davies, and Alan L. Parker. 2015. "Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications" Viruses 7, no. 11: 6009-6042. https://doi.org/10.3390/v7112923
APA StyleUusi-Kerttula, H., Hulin-Curtis, S., Davies, J., & Parker, A. L. (2015). Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications. Viruses, 7(11), 6009-6042. https://doi.org/10.3390/v7112923