
Psoralen Inactivation of Viruses: A Process for the Safe Manipulation of Viral Antigen and Nucleic Acid

Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Propagation
2.2. Virus Inactivation by Psoralen
2.3. TCID50
2.4. Antibody Production
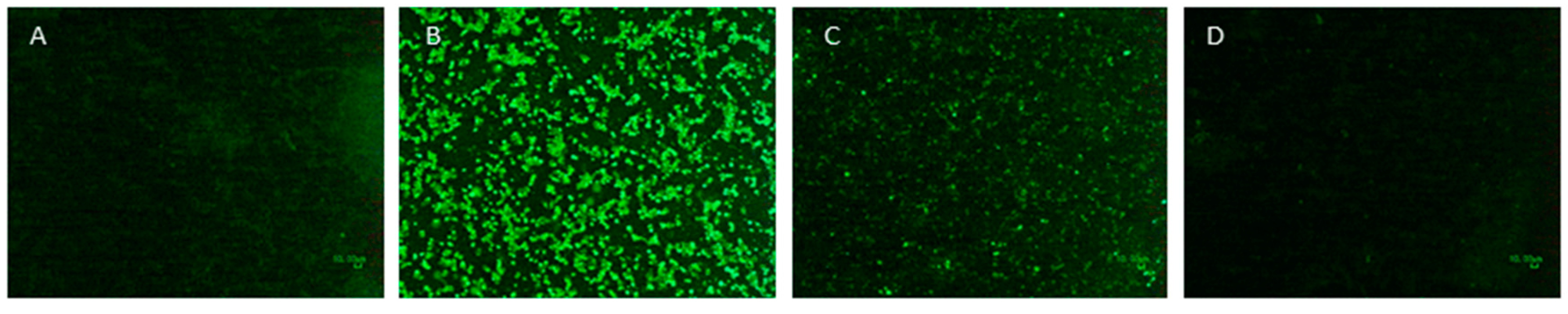
2.5. Immunofluorescence Assay (IFA)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Primary Antibody | Primary Source | Secondary Antibody | Secondary Source |
|---|---|---|---|---|
| VEEV | Mouse monoclonal | Millipore 1 | Goat anti-mouse | KPL 6 |
| JUNV | Ascitic fluid | ATCC 2 | Goat anti-mouse | KPL |
| RVFV | Rabbit polyclonal | NBACC 3 | Goat anti-rabbit | KPL |
| MERS-CoV | Rabbit polyclonal | Sino Biological 4 | Goat anti-rabbit | KPL |
| EBOV | Rabbit polyclonal | NBACC | Goat anti-rabbit | KPL |
| DENV | Mouse monoclonal | GenWay Biotech 5 | Goat anti-mouse | KPL |
2.6. Flow Cytometry
2.7. Oligonucleotide Primer and Probe Design and Generation
| Name | Target | Sequence | Product Size (bp) | Reference |
|---|---|---|---|---|
| DENV-8 F | NS3 | GCTGAAATGGAGGAAGCCCT | 637 | this study |
| DENV-8 R | CCCGCTCTTCACCATCTGTT | |||
| DENV-8 P | CAGAGCTGAGCACACCGGGC | |||
| DENV-9 F | NS4B | AGTTCCCCTTCTCGCCATTG | 961 | this study |
| DENV-9 R | GAGTGTTCGTCCTGCTTCCA | |||
| DENV-9 P | AAAAGAGCAGCGGCGGGCAT | |||
| DENV-3 F | RdRp/NS5 | TACAACATGATGGGAAAGCGAGAGAAAAA | 265 | [28] |
| DENV-3 R | GTGTCCCAGCCGGCGGTGTCATCAGC | |||
| DENV-3 P | AAGAGACGTGAGCAGGAAGGAAGGGGGAGC | |||
| DENV-10 F | NS5 | GGAGGAGCAATGTATGCCGA | 735 | this study |
| DENV-10 R | GTCGCGTCTGTGGAAGTACA | |||
| DENV-10 P | GGTCTTTGCGGGAGACGGCC | |||
| DENV-11 F | NS5/UTR | AGAGAAGACCAATGGTGCGG | 488 | this study |
| DENV-11 R | CCTTCCAGCGAGACTACAGC | |||
| DENV-11 P | CTACCTGTGAGCCCCGTCCAAG | |||
| EBOV-Z F | NP | TGGAAAAAACATTAAGAGAACACTTGC | 79 | [29] |
| EBOV-Z R | AGGAGAGAAACTGACCGGCAT | |||
| EBOV-Z P | CATGCCGGAAGAGGAGACAACTGAAGC | |||
| JUNV F | NP | CAGTTCATCCCTCCCCAGATC | 79 | [30] |
| JUNV R | GGTTGACAGACTTATGTCCATGAAGA | |||
| JUNV P | TGTTCAACGAAACACAGTTTTCAAGGTGGG | |||
| LASV F | GPC | TGCTAGTACAGACAGTGCAATGAG | 79 | [30] |
| LASV R | TAGTGACATTCTTCCAGGAAGTGC | |||
| LASV P | TGTTCATCACCTCTTC | |||
| MARV F | VP40 | GGACCACTGCTGGCCATATC | 103 | [31] |
| MARV R | GAGAACATITCGGCAGGAAG | |||
| MARV P | ATCCTAAACAGGCTTGTCTTCTCTGGGAC TT | |||
| RVFV F | L | TGAAAATTCCTGAGACACATGG | 89 | [32] |
| RVFV R | ACTTCCTTGCATCATCTGATG | |||
| RVFV P | CAATGTAAGGGGCCTGTGTGGACTTGTG |
2.8. RNA Extraction
2.9. Reverse Transcription Real-Time PCR Assay
2.10. Sanger Sequencing
2.11. Next Generation Sequencing
3. Results
3.1. Inactivation of DENV Using a Psoralen and UV-A
| Minutes UV-A Exposure | UV-A µW/cm2 | Passage 1 | Passage 2 | |||
|---|---|---|---|---|---|---|
| Log10 TCID50/mL | CPE | IFA | CPE | IFA | ||
| 0 | 0 | 6.4 | yes | yes | yes | yes |
| 2 | 400 | 5.8 | yes | yes | yes | yes |
| 5 | 1000 | 4.9 | yes | yes | yes | yes |
| 10 | 2000 | 3.5 | yes | yes | yes | yes |
| 20 | 4000 | 1.9 | yes | yes | yes | yes |
| 30 | 6000 | 0 | no | no | yes | yes |
| 40 | 8000 | 0 | no | no | no | no |
| 50 | 10,000 | 0 | no | no | no | no |
| 60 | 12,000 | 0 | no | no | no | no |
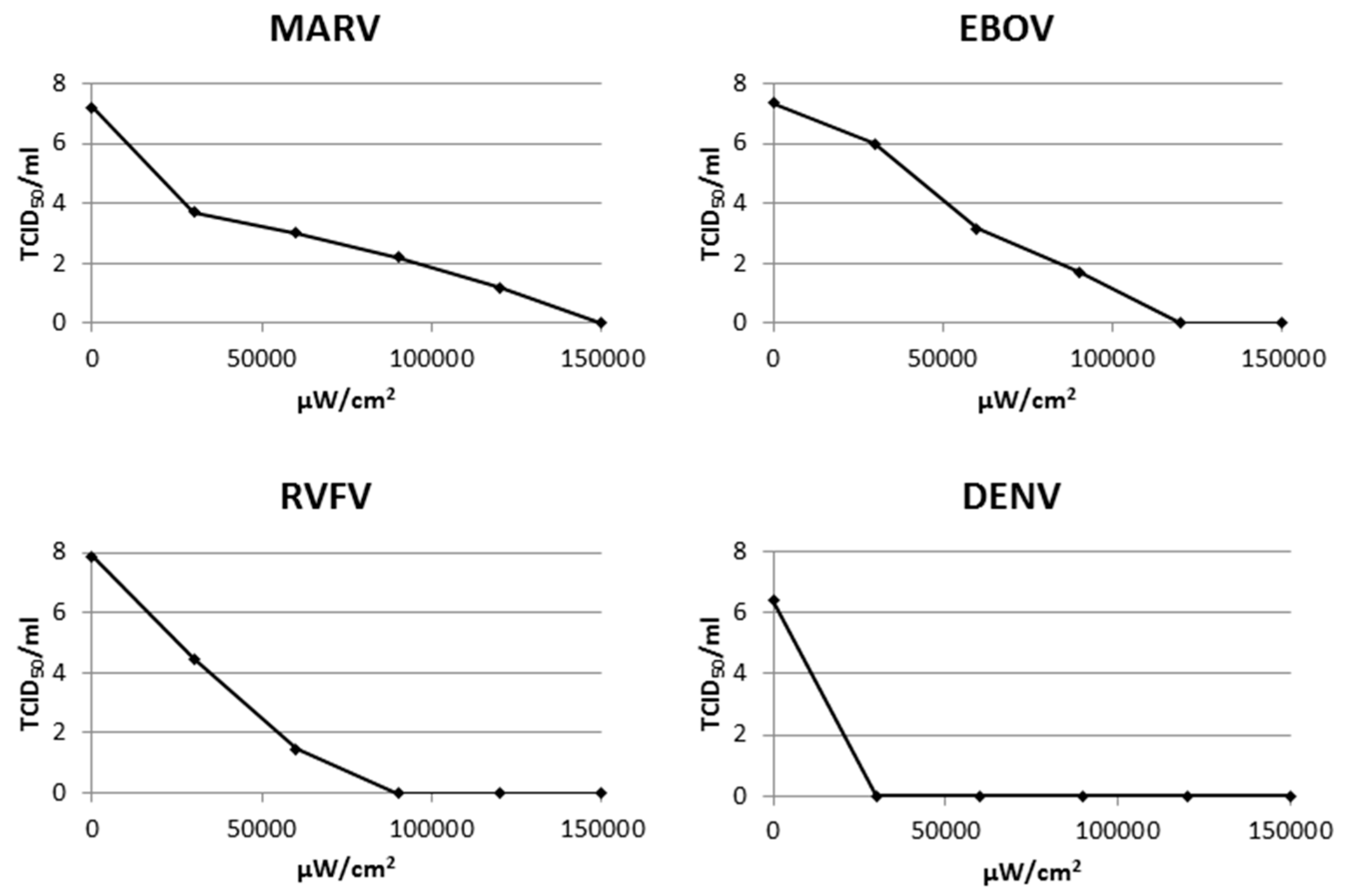
3.2. Inactivation of Multiple Additional Viruses Using a Psoralen and UV-A
| Family | Characteristics | Virus | UV-A + AMT Required for Inactivation (µW/cm2) |
|---|---|---|---|
| Alphaviridae | enveloped, spherical, ssRNA(+) | VEEV | 20,000 |
| Arenaviridae | enveloped, spherical, ssRNA(-) | LASV | 30,000 |
| JUNV | 90,000 | ||
| Bunyaviridae | enveloped, spherical, ssRNA(-) | RVFV | 90,000 |
| CCHFV | 4000 | ||
| Coronaviridae | enveloped, spherical, ssRNA(+) | MERS-CoV | 6000 |
| Filoviridae | filamentous, ssRNA(-) | MARV | 150,000 |
| EBOV | 120,000 | ||
| Flaviviridae | enveloped, spherical, ssRNA(+) | DENV | 8000 |
| WNV | 2000 | ||
| SLEV | 1000 | ||
| YFV | 2000 | ||
| Orthomyxoviridae | enveloped, usually spherical but can be filamentous, ssRNA(-) | H1N1p | 1000 |
| H1N1 | 1000 | ||
| H3N2 | 2000 | ||
| Flu-B | 2000 |
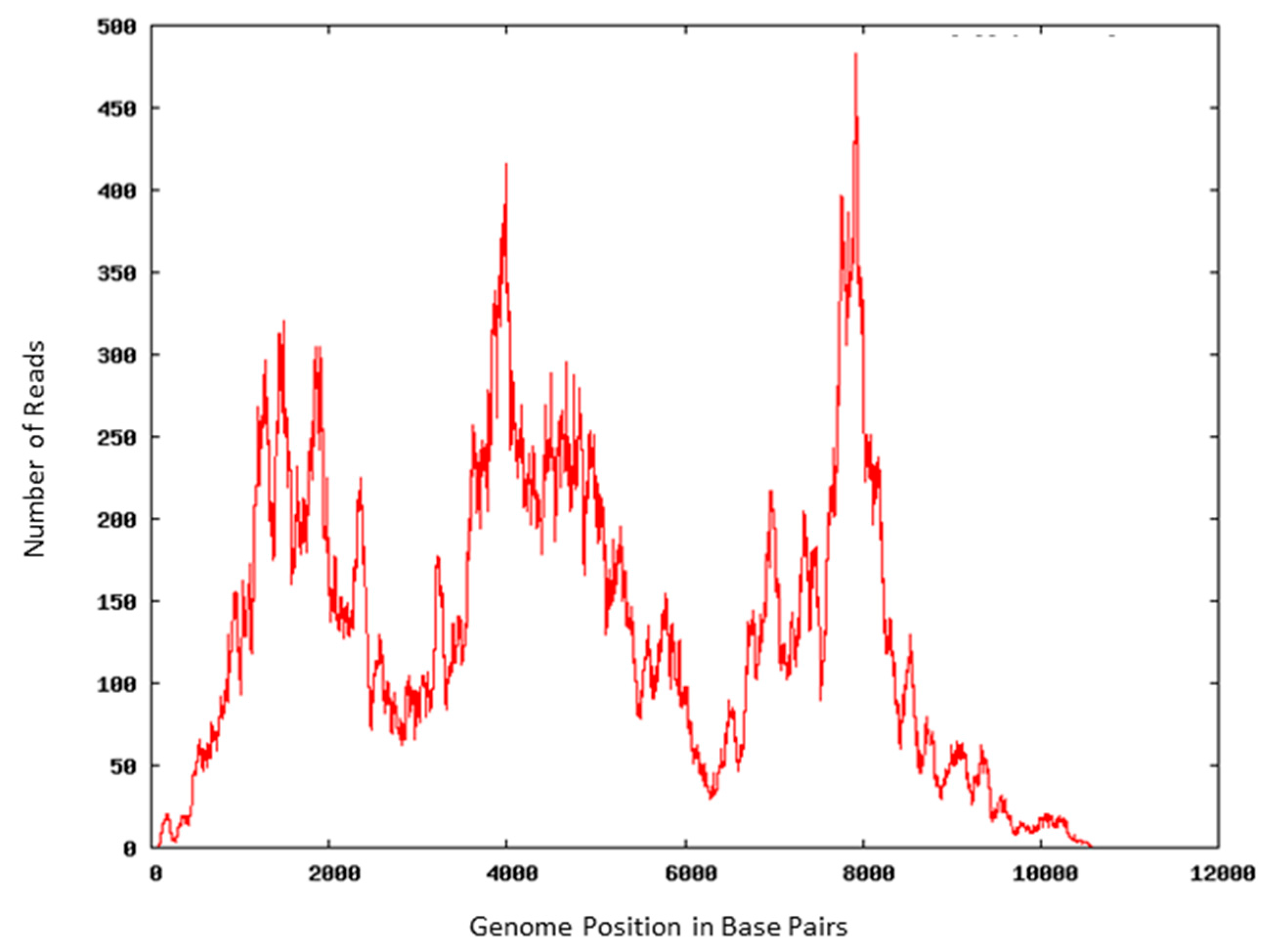
3.3. Viral RNA Retains Molecular Integrity after AMT Treatment
| Primers | Target | Size (base pairs) | Nucleotide Location | Average Ct/Minutes AMT + UV-A Exposure | ||
|---|---|---|---|---|---|---|
| 0 | 30 | 60 | ||||
| DENV-8 | NS3 | 637 | 5206 | 15.39 | 17.53 | 19.30 |
| DENV-9 | NS4B | 961 | 7095 | 17.32 | 20.22 | 20.21 |
| DENV-3 | RdRp/NS5 | 120 | 8923 | 28.63 | 29.38 | 30.31 |
| DENV-10 | NS5 | 735 | 9148 | 21.10 | 22.70 | 21.35 |
| DENV-11 | NS5/UTR | 489 | 10093 | 17.01 | 17.25 | 18.77 |
3.4. Viral Surface Epitopes Maintain Integrity after Psoralen Treatment
| Agent | Rabbit Number | Titer |
|---|---|---|
| AMT + UV-A LASV | 479876 | 1:320 |
| AMT + UV-A LASV | 479877 | 1:320 |
| AMT + UV-A LASV | 479878 | 1:320 |
| LASV | 479879 | 1:320 |
| LASV | 479880 | 1:320 |
| LASV | 479881 | 1:640 |
| AMT + UV-A CCHFV | 479882 | 1:160 |
| AMT + UV-A CCHFV | 479883 | 1:320 |
| AMT + UV-A CCHFV | 479884 | 1:640 |
| CCHFV | 479885 | 1:320 |
| CCHFV | 479886 | 1:320 |
| CCHFV | 479887 | 1:320 |
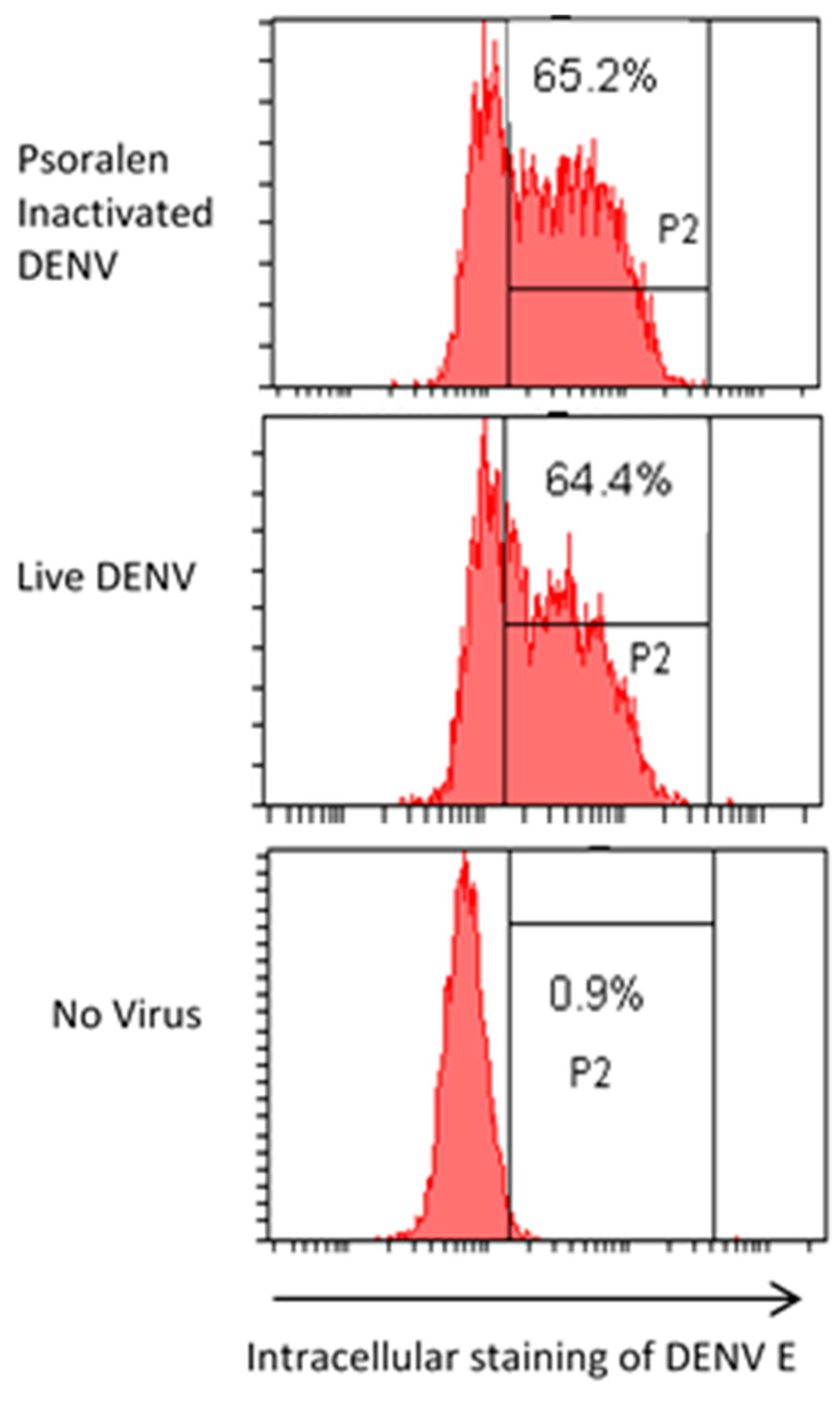
3.5. AMT + UV-A Treated Virus Loses Infectivity While Retaining Receptor Binding
3.6. AMT-Inactivated Viruses Specifically Bind Cell Receptors
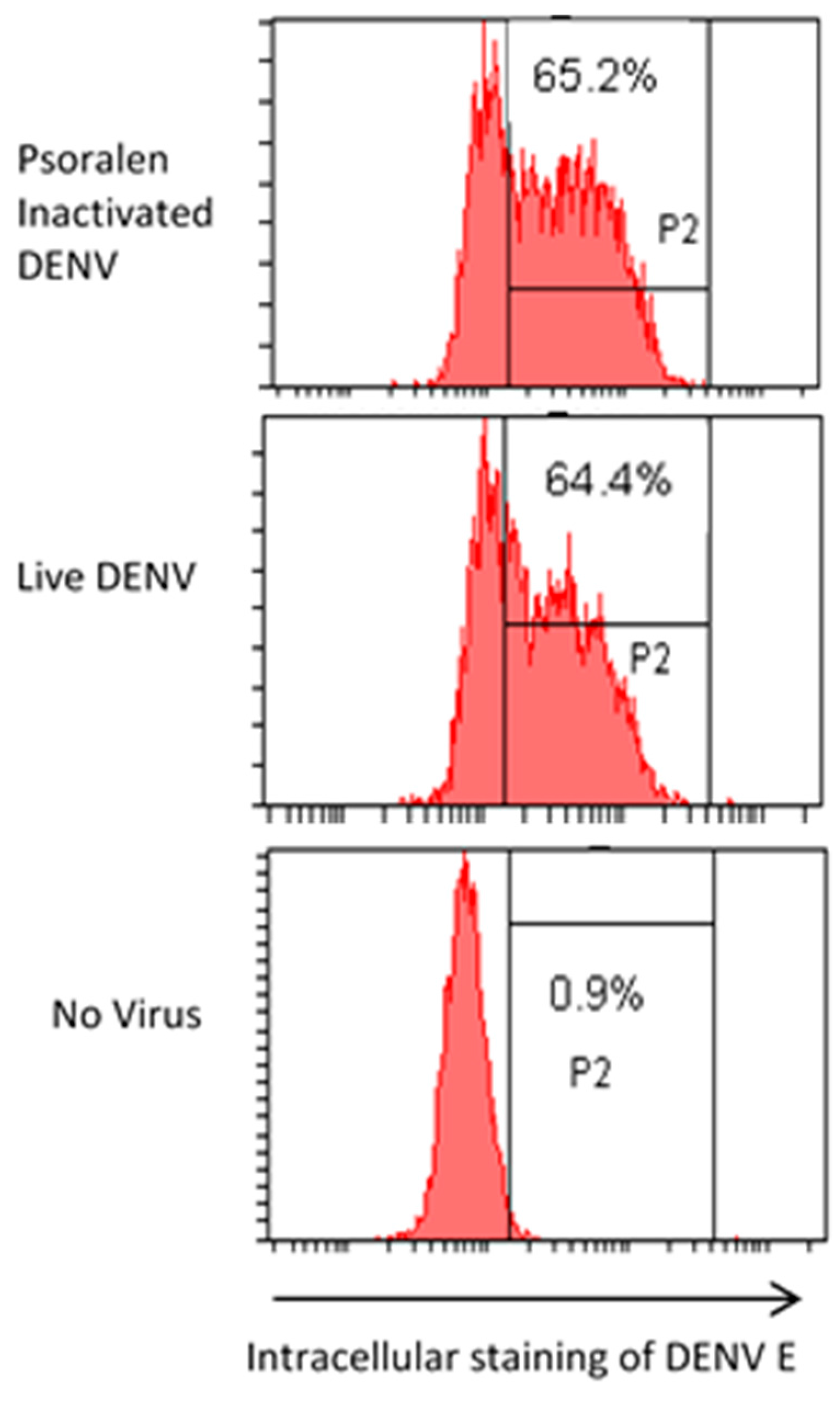
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sagripanti, J.L.; Hülseweh, B.; Grote, G.; Voss, L.; Böhling, K.; Marschall, H.J. Microbial inactivation for safe and rapid diagnostics of infectious samples. Appl. Environ. Microbiol. 2011, 77, 7289–7295. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, N.B.; Nguyen, H.V.; Winters, C.A.; Brantner, C.A.; Smith, C.L.; Andrews, S.B. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J. Neurosci. 2004, 24, 5611–5622. [Google Scholar] [CrossRef] [PubMed]
- Moller, L.; Schunadel, L.; Nitsche, A.; Schwebke, I.; Hanisch, M.; Laue, M. Evaluation of virus inactivation by formaldehyde to enhance biosafety of diagnostic electron microscopy. Viruses 2015, 7, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Kading, R.; Crabtree, M.; Miller, B. Inactivation of infectious virus and serological detection of virus antigen in Rift Valley fever virus-exposed mosquitoes fixed with paraformaldehyde. J. Virol. Methods 2013, 189, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Wilton, T.; Dunn, G.; Eastwood, D.; Minor, P.D.; Martin, J. Effect of formaldehyde inactivation on poliovirus. J. Virol. 2014, 88, 11955–11964. [Google Scholar] [CrossRef] [PubMed]
- She, Y.M.; Cheng, K.; Farnsworth, A.; Li, X.; Cyr, T.D. Surface modifications of influenza proteins upon virus inactivation by β-propiolactone. Proteomics 2013, 13, 3537–3547. [Google Scholar] [CrossRef] [PubMed]
- Delrue, I.; Delputte, P.L.; Nauwynck, H.J. Assessing the functionality of viral entry-associated domains of porcine reproductive and respiratory syndrome virus during inactivation procedures, a potential tool to optimize inactivated vaccines. Vet. Res. 2009, 40, 62. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Blow, J.A.; Dohm, D.J.; Negley, D.L.; Mores, C.N. Virus inactivation by nucleic acid extraction reagents. J. Virol. Methods 2004, 119, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Raviprakash, K.; Sun, P.; Raviv, Y.; Luke, T.; Martin, N.; Kochel, T. Dengue virus photo-inactivated in presence of 1,5-iodonaphthylazide (INA) or AMT, a psoralen compound (4′-aminomethyl-trioxsalen) is highly immunogenic in mice. Hum. Vaccines Immunother. 2013, 9, 2336–2341. [Google Scholar] [CrossRef]
- Groene, W.S.; Shaw, R.D. Psoralen preparation of antigenically intact noninfectious rotavirus particles. J. Virol. Methods 1992, 38, 93–102. [Google Scholar] [CrossRef]
- Hanson, C.V.; Riggs, J.L.; Lennette, E.H. Photochemical inactivation of DNA and RNA viruses by psoralen derivatives. J. Gen. Virol. 1978, 40, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Allain, J.P.; Hsu, J.; Pranmeth, M.; Hanson, D.; Stassinopoulos, A.; Fischetti, L.; Corash, L.; Lin, L. Quantification of viral inactivation by photochemical treatment with amotosalen and UV A light, using a novel polymerase chain reaction inhibition method with preamplification. J. Infect. Dis. 2006, 194, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cook, D.N.; Wiesehahn, G.P.; Alfonso, R.; Behrman, B.; Cimino, G.D.; Corten, L.; Damonte, P.B.; Dikeman, R.; Dupuis, K.; et al. Photochemical inactivation of viruses and bacteria in platelet concentrates by use of a novel psoralen and long-wavelength ultraviolet light. Transfusion 1997, 37, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.L.; Dodd, R.Y. Reducing the risk of blood transfusion. Hematol. Am. Soc. Hematol. Educ. Program 2001, 1, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.; Ros, A.M. Treatment of psoriasis with psoralens and ultraviolet A. A double-blind comparison of 8-methoxypsoralen and 5-methoxypsoralen. Photodermatol. Photoimmunol. Photomed. 1994, 10, 217–220. [Google Scholar] [PubMed]
- McNeal, M.M.; Rae, M.N.; Ward, R.L. Effects of different adjuvants on rotavirus antibody responses and protection in mice following intramuscular immunization with inactivated rotavirus. Vaccine 1999, 17, 1573–1580. [Google Scholar] [CrossRef]
- Tang, J.; Murtadha, M.; Schnell, M.; Eisenlohr, L.C.; Hooper, J.; Flomenberg, P. Human t-cell responses to vaccinia virus envelope proteins. J. Virol. 2006, 80, 10010–10020. [Google Scholar] [CrossRef] [PubMed]
- Sutjipto, S.; Pedersen, N.C.; Miller, C.J.; Gardner, M.B.; Hanson, C.V.; Gettie, A.; Jennings, M.; Higgins, J.; Marx, P.A. Inactivated simian immunodeficiency virus vaccine failed to protect rhesus macaques from intravenous or genital mucosal infection but delayed disease in intravenously exposed animals. J. Virol. 1990, 64, 2290–2297. [Google Scholar] [PubMed]
- Kochel, T.J.; Maves, R.C.; Porter, K. A Psoralen-Inactivated Dengue Virus Vaccine and Method of Preparation. U.S. Patent 9,005,633 B2, 14 April 2015. [Google Scholar]
- Maves, R.C.; Castillo Oré, R.M.; Porter, K.R.; Kochel, T.J. Immunogenicity of a psoralen-inactivated Dengue virus type 1 vaccine candidate in mice. Clin. Vaccine Immunol. 2010, 17, 304–306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maves, R.C.; Oré, R.M.; Porter, K.R.; Kochel, T.J. Immunogenicity and protective efficacy of a psoralen-inactivated Dengue-1 virus vaccine candidate in Aotus nancymaae monkeys. Vaccine 2011, 29, 2691–2696. [Google Scholar] [CrossRef] [PubMed]
- Lennette, D.A. General principles for laboratory diagnosis of viral, rickettsial, and chlamydial infections. In Diagnostic Procedure for Viral, Rickettsial, and Chlamydial Infections, 7th ed.; Lennette, E.H., Lennette, D.A., Lennette, E.T., Eds.; American Public Health Association: Washington, DC, USA, 1995; pp. 3–25. [Google Scholar]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- Hsiung, G.D. Virus isolation and identification methods. In Hsiung’s Diagnostic Virology, 4th ed.; Yale University Press: New Haven, CT, USA, 1994; pp. 37–45. [Google Scholar]
- Hsiung, G.D. Immunocytochemical assays: Immunofluorescent and immunoperoxidase staining for antigen detection. In Hsiung’s Diagnostic Virology, 4th ed.; Yale University Press: New Haven, CT, USA, 1994; pp. 56–61. [Google Scholar]
- Wu, L.; Martin, T.D.; Carrington, M.; KewalRamani, V.N. Raji B cells, misidentified as THP-1 cells, stimulate DC-SIGN-mediated HIV transmission. Virology 2004, 318, 17–23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chien, L.J.; Liao, T.L.; Shu, P.Y.; Huang, J.H.; Gubler, D.; Chang, G.J.J. Development of real-time reverse transcriptase PCR assays to detect and serotype Dengue viruses. J. Clin. Microbiol. 2006, 44, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Sealy, T.K.; Ksiazek, T.G.; Nichol, S.T. High-throughput molecular detection of hemorrhagic fever virus threats with applications for outbreak settings. J. Infect. Dis. 2007, 196 (Suppl. 2), S205–S212. [Google Scholar] [CrossRef] [PubMed]
- Trombley, A.R.; Wachter, L.; Garrison, J.; Buckley-Beason, V.A.; Jahrling, J.; Hensley, L.E.; Schoepp, R.J.; Norwood, D.A.; Goba, A.; Fair, J.N.; et al. Comprehensive panel of real-time taqman™ polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and new world hantaviruses. Am. J. Trop. Med. Hyg. 2010, 82, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Ströher, U.; et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Bawiec, D.A.; Ksiazek, T.G.; Shoemaker, T.R.; Nichol, S.T. Highly sensitive and broadly reactive quantitative reverse transcription-PCR assay for high-throughput detection of Rift Valley fever virus. J. Clin. Microbiol. 2007, 45, 3506–3513. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Paired-end Sequencing. Available online: http://www.illumina.com/technology/next-generation-sequencing/paired-end-sequencing_assay.html (accessed on 4 November 2015).
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of Dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef] [PubMed]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L.; et al. DC-SIGN (CD209) mediates Dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, K.; Wronka-Edwards, L.; Leggett-Embrey, M.; Walker, E.; Sun, P.; Ondov, B.; Wyman, T.H.; Rosovitz, M.; Bohn, S.S.; Burans, J.; et al. Psoralen Inactivation of Viruses: A Process for the Safe Manipulation of Viral Antigen and Nucleic Acid. Viruses 2015, 7, 5875-5888. https://doi.org/10.3390/v7112912
Schneider K, Wronka-Edwards L, Leggett-Embrey M, Walker E, Sun P, Ondov B, Wyman TH, Rosovitz M, Bohn SS, Burans J, et al. Psoralen Inactivation of Viruses: A Process for the Safe Manipulation of Viral Antigen and Nucleic Acid. Viruses. 2015; 7(11):5875-5888. https://doi.org/10.3390/v7112912
Chicago/Turabian StyleSchneider, Katherine, Loni Wronka-Edwards, Melissa Leggett-Embrey, Eric Walker, Peifang Sun, Brian Ondov, Travis H. Wyman, MJ Rosovitz, Sherry S. Bohn, James Burans, and et al. 2015. "Psoralen Inactivation of Viruses: A Process for the Safe Manipulation of Viral Antigen and Nucleic Acid" Viruses 7, no. 11: 5875-5888. https://doi.org/10.3390/v7112912
APA StyleSchneider, K., Wronka-Edwards, L., Leggett-Embrey, M., Walker, E., Sun, P., Ondov, B., Wyman, T. H., Rosovitz, M., Bohn, S. S., Burans, J., & Kochel, T. (2015). Psoralen Inactivation of Viruses: A Process for the Safe Manipulation of Viral Antigen and Nucleic Acid. Viruses, 7(11), 5875-5888. https://doi.org/10.3390/v7112912