Super-Resolution Microscopy: A Virus’ Eye View of the Cell

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
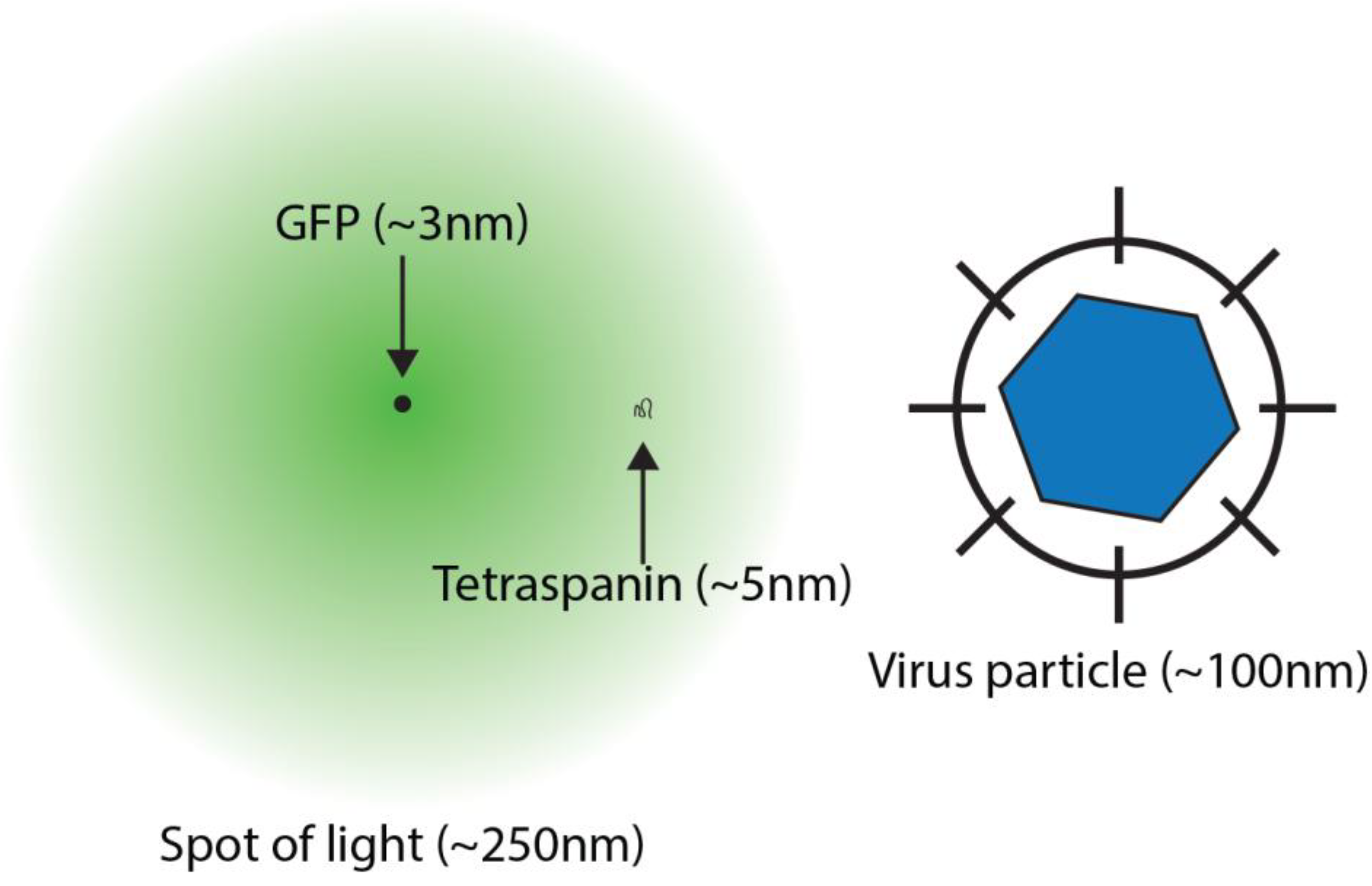
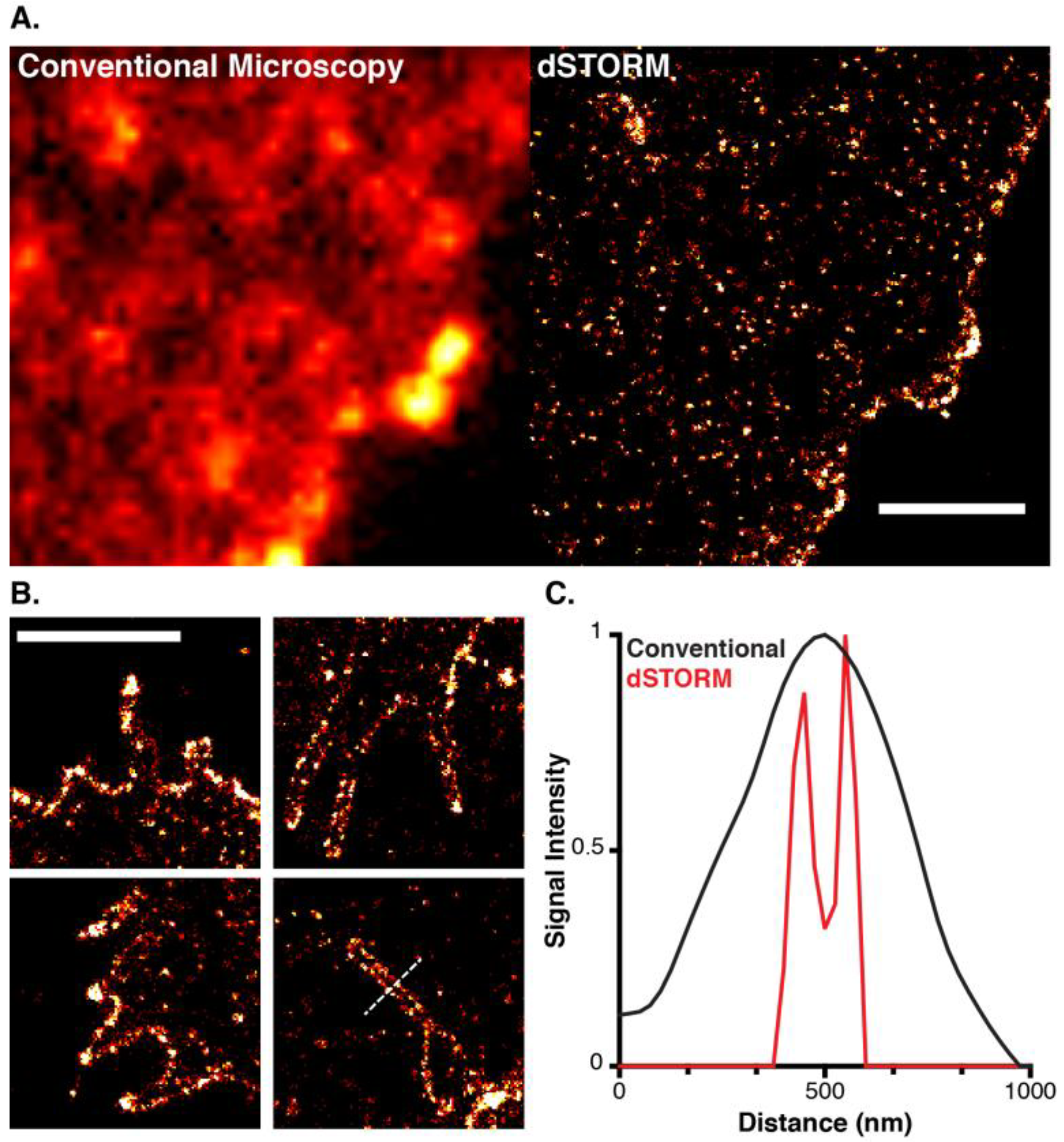
2. Super-Resolution Imaging
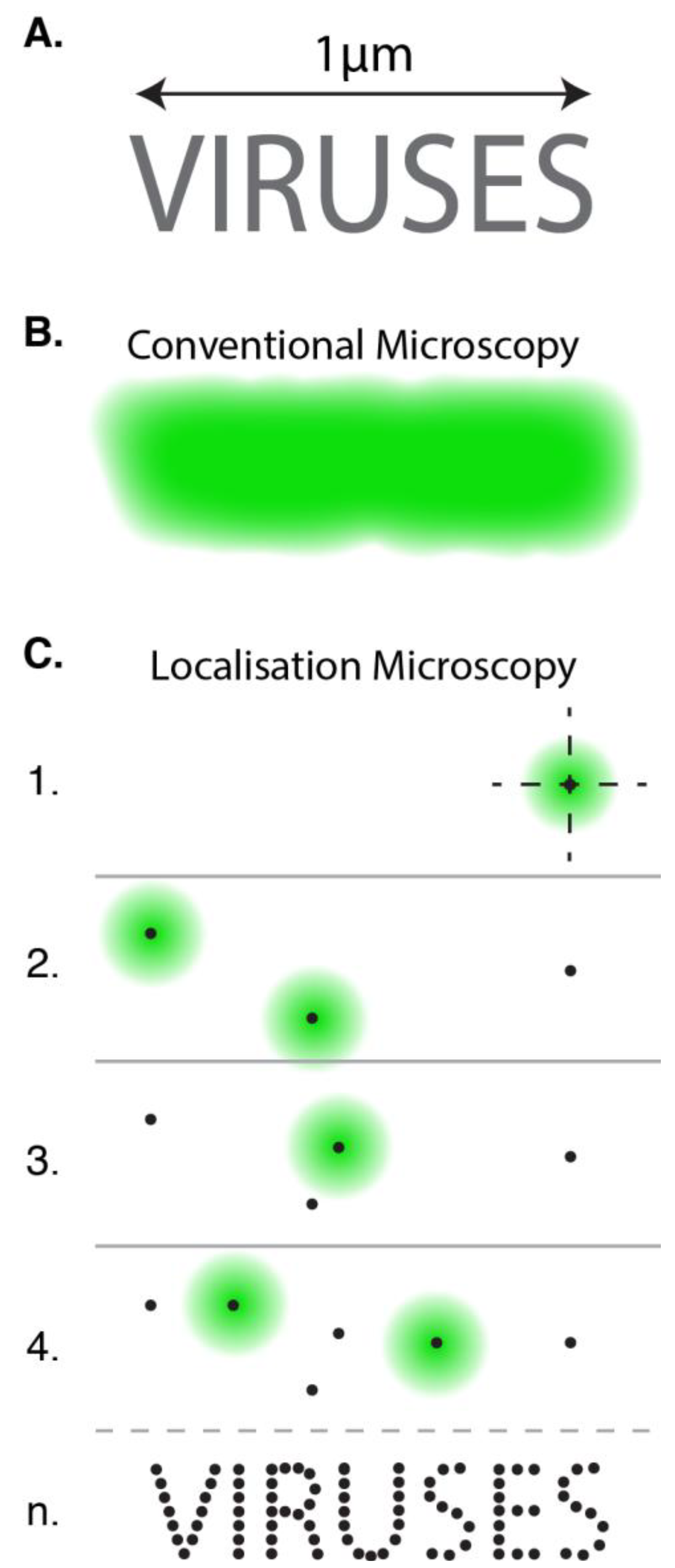
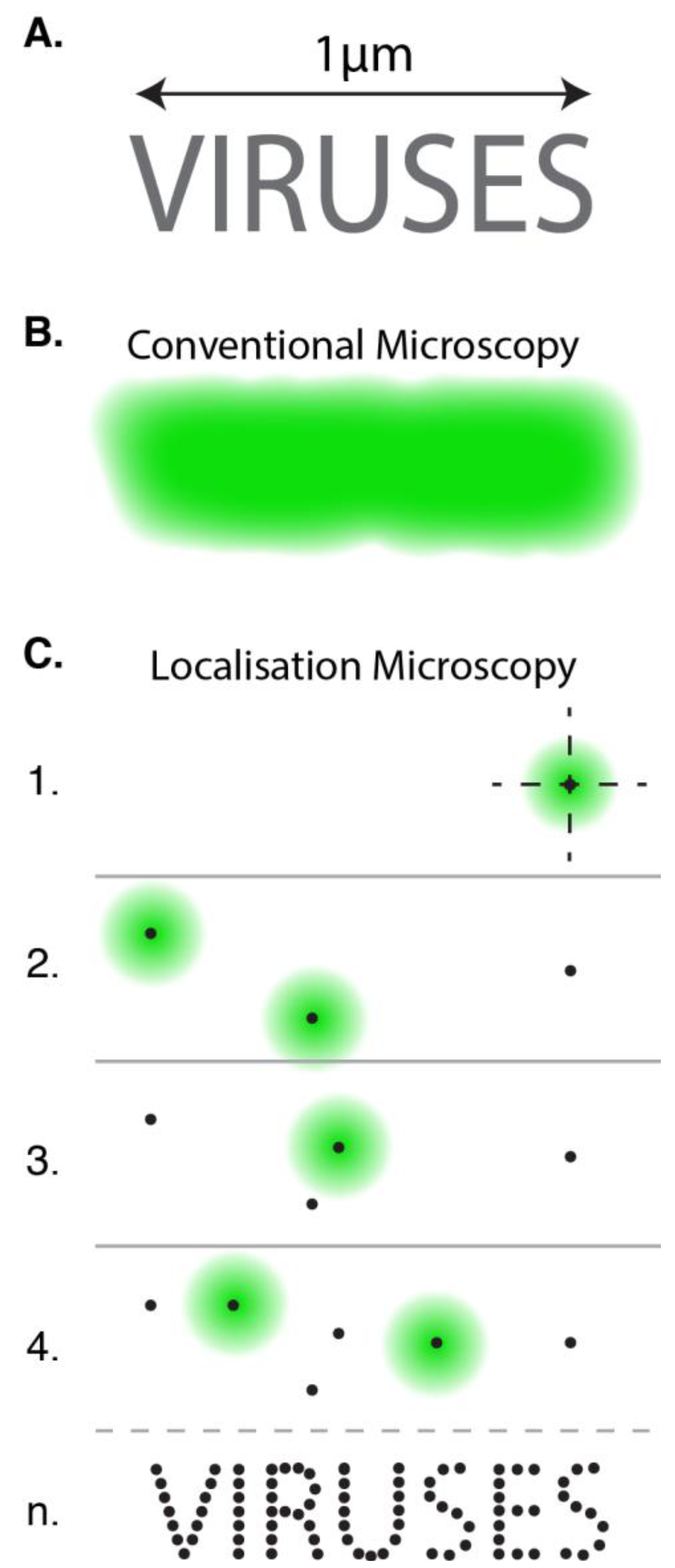
3. Localisation Microscopy
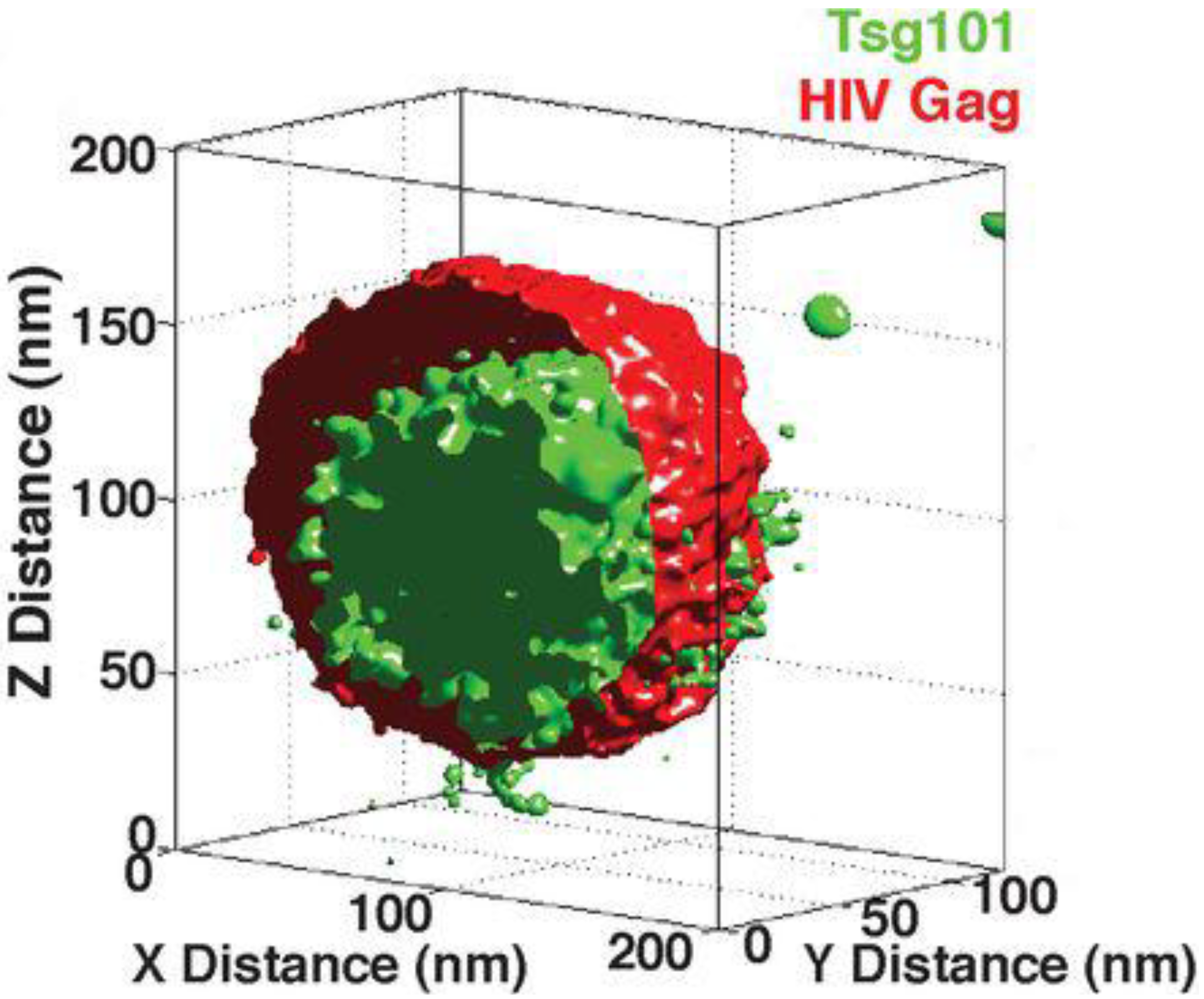
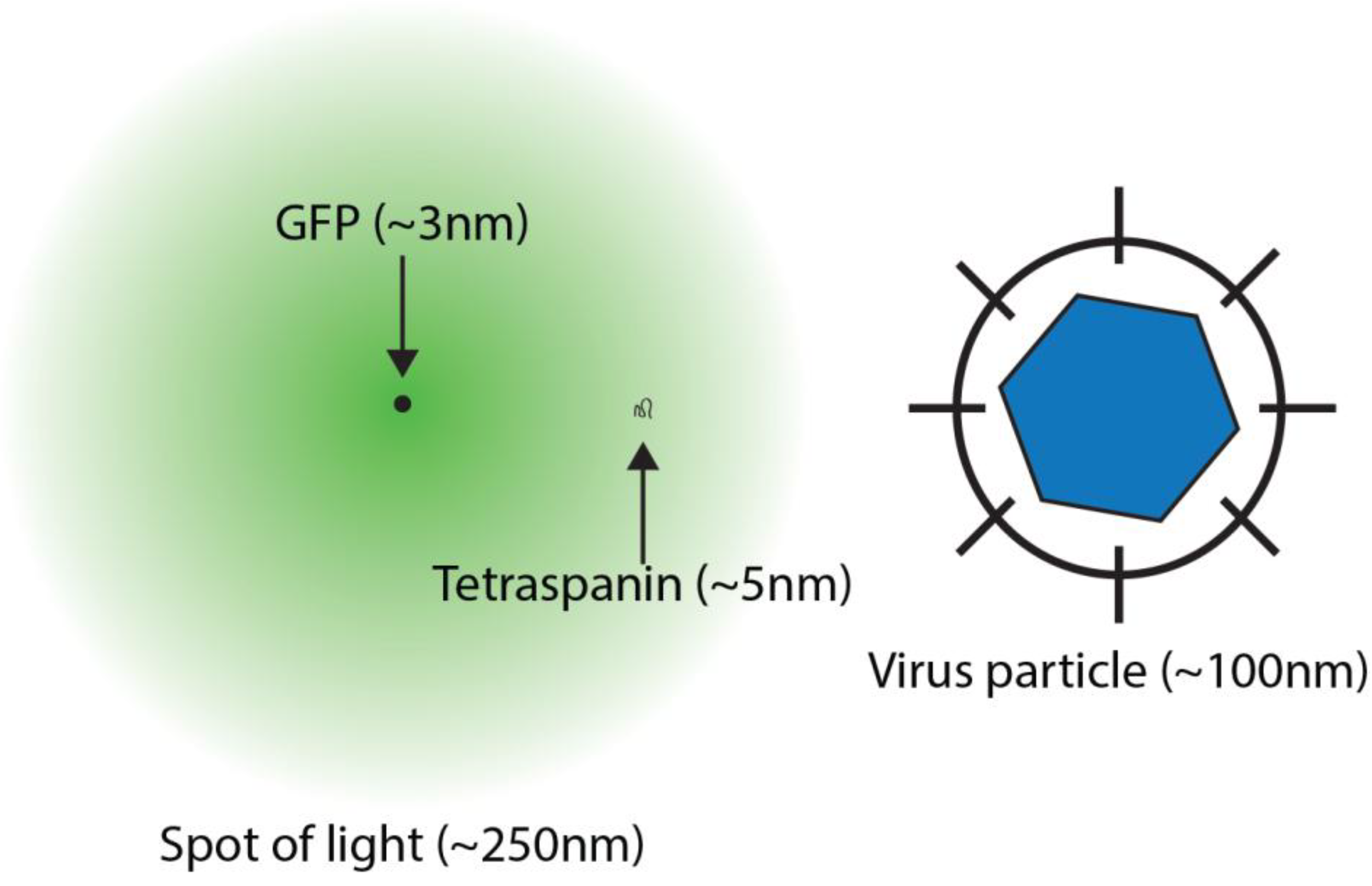
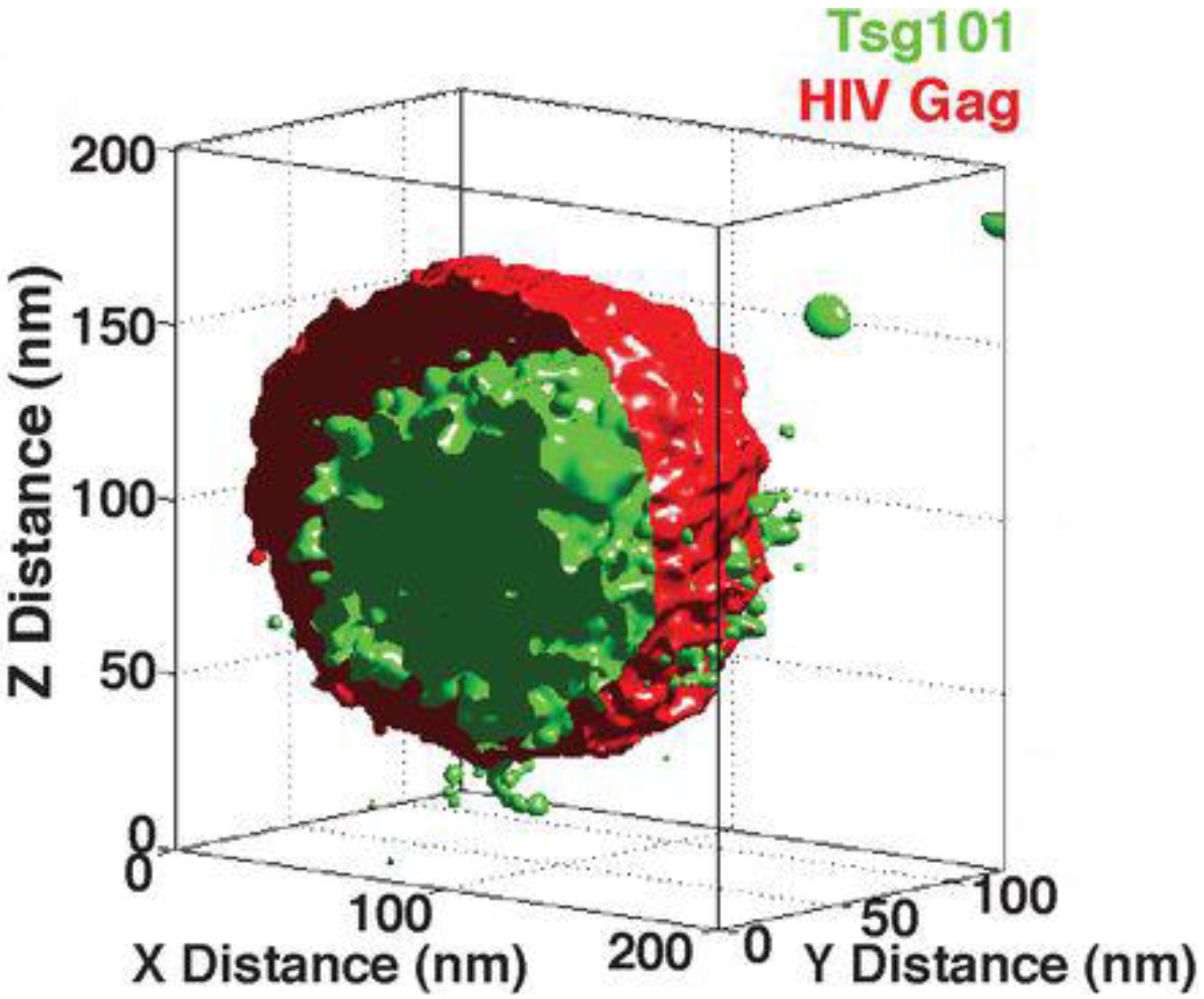
4. Super-Resolved Viruses
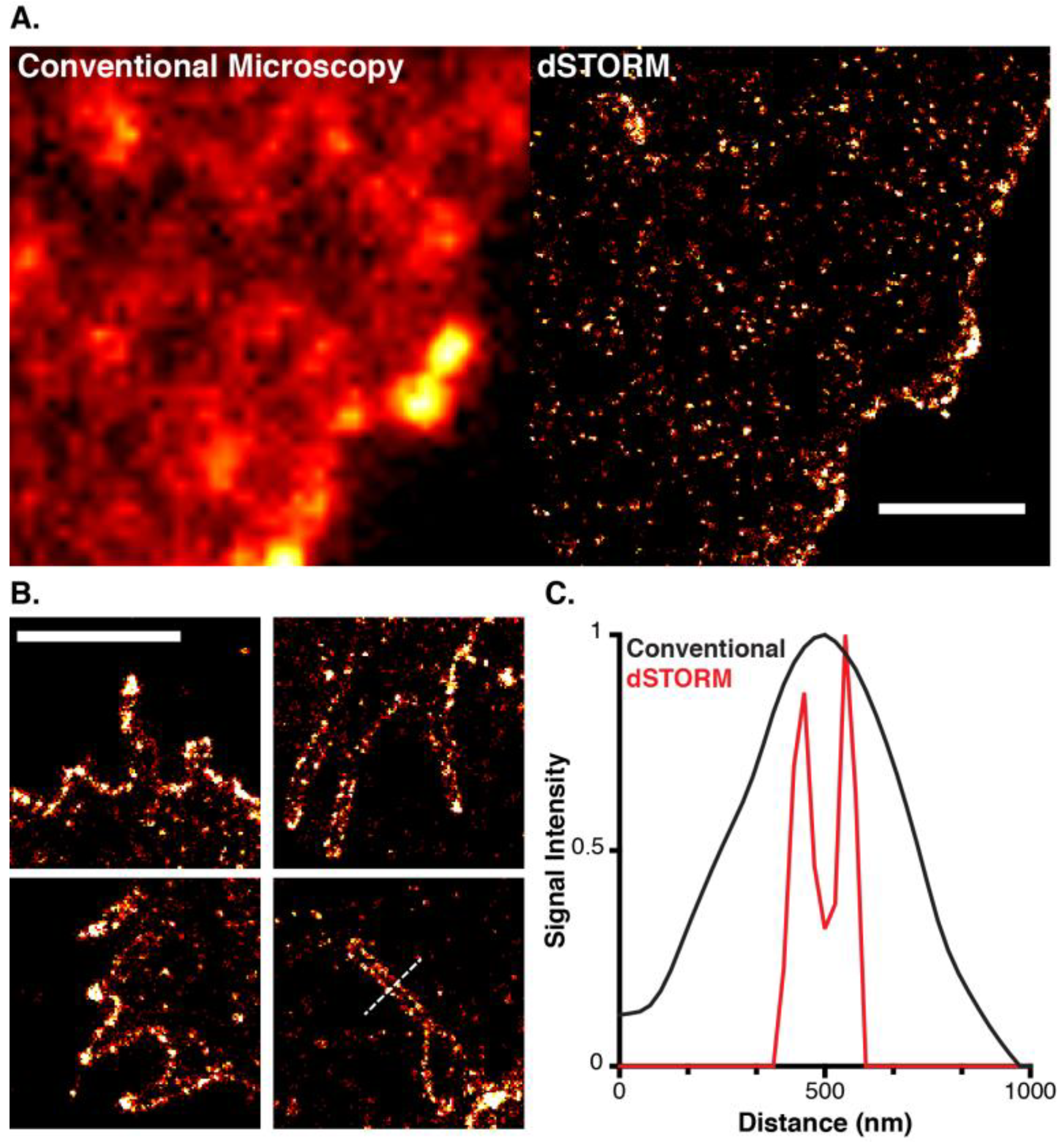
5. Observing Tetraspanin Biology
6. A Virus’ Eye View of CD81
7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- De Jonge, N.; Peckys, D.B.; Kremers, G.J.; Piston, D.W. Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 2159–2164. [Google Scholar]
- Yildiz, A.; Forkey, J.N.; Mckinney, S.A.; Ha, T.; Goldman, Y.E.; Selvin, P.R. Myosin V walks hand-over-hand: Single fluorophore imaging with 1.5-nm localization. Science 2003, 300, 2061–2065. [Google Scholar] [CrossRef]
- Espenel, C.; Margeat, E.; Dosset, P.; Arduise, C.; le Grimellec, C.; Royer, C.A.; Boucheix, C.; Rubinstein, E.; Milhiet, P.-E. Single-molecule analysis of CD9 dynamics and partitioning reveals multiple modes of interaction in the tetraspanin web. J. Cell Biol. 2008, 182, 765–776. [Google Scholar] [CrossRef]
- Abbe, E. Contributions to the theory of the microscope and microscopic detection. (translated from German). Arch. Mikrosk. Anat. 1873, 9, 413–418. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Meth. 2006, 3, 793–795. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Hess, S.T.; Girirajan, T.P.K.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef]
- Annibale, P.; Vanni, S.; Scarselli, M.; Rothlisberger, U.; Radenovic, A. Quantitative photo activated localization microscopy: Unraveling the effects of photoblinking. PLoS One 2011, 6, e22678. [Google Scholar]
- Lee, S.-H.; Shin, J.Y.; Lee, A.; Bustamante, C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM). Proc. Natl. Acad. Sci. USA 2012, 109, 17436–17441. [Google Scholar] [CrossRef]
- Puchner, E.M.; Walter, J.M.; Kasper, R.; Huang, B.; Lim, W.A. Counting molecules in single organelles with superresolution microscopy allows tracking of the endosome maturation trajectory. Proc. Natl. Acad. Sci. USA 2013, 110, 16015–16020. [Google Scholar]
- Heilemann, M.; van de Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008, 47, 6172–6176. [Google Scholar] [CrossRef]
- Nyquist, H. Certain topics in telegraph transmission theory. (Reprinted from Transactions of the American Institute of Electrical Engineers, 1928, 47, 617–644). Proc. IEEE 2002, 90, 280–305. [Google Scholar] [CrossRef]
- Van de Linde, S.; Sauer, M. How to switch a fluorophore: From undesired blinking to controlled photoswitching. Chem. Soc. Rev. 2014, 43, 1076–1087. [Google Scholar] [CrossRef]
- Nienhaus, K.; Ulrich Nienhaus, G. Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev. 2014, 43, 1088–1106. [Google Scholar] [CrossRef]
- Sengupta, P.; van Engelenburg, S.B.; Lippincott-Schwartz, J. Superresolution imaging of biological systems using photoactivated localization microscopy. Chem. Rev. 2014. [Google Scholar] [CrossRef]
- Sengupta, P.; Jovanovic-Talisman, T.; Lippincott-Schwartz, J. Quantifying spatial organization in point-localization superresolution images using pair correlation analysis. Nat. Protoc. 2013, 8, 345–354. [Google Scholar] [CrossRef]
- Metcalf, D.J.; Edwards, R.; Kumarswami, N.; Knight, A.E. Test samples for optimizing STORM super-resolution microscopy. J. Vis. Exp. 2013. [Google Scholar] [CrossRef]
- Curthoys, N.M.; Mlodzianoski, M.J.; Kim, D.; Hess, S.T. Simultaneous multicolor imaging of biological structures with fluorescence photoactivation localization microscopy. J. Vis. Exp. 2013, e50680. [Google Scholar]
- Allen, J.R.; Ross, S.T.; Davidson, M.W. Sample preparation for single molecule localization microscopy. Phys. Chem. Chem. Phys. 2013, 15, 18771–18783. [Google Scholar] [CrossRef]
- Huang, F.; Hartwich, T.M.P.; Rivera-Molina, F.E.; Lin, Y.; Duim, W.C.; Long, J.J.; Uchil, P.D.; Myers, J.R.; Baird, M.A.; Mothes, W.; et al. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nat. Meth. 2013, 10, 653–658. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nature 2010, 468, 580–584. [Google Scholar] [CrossRef]
- Jones, S.A.; Shim, S.-H.; He, J.; Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Meth. 2011, 8, 499–508. [Google Scholar] [CrossRef]
- Lelek, M.; di Nunzio, F.; Henriques, R.; Charneau, P.; Arhel, N.; Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. USA 2012, 109, 8564–8569. [Google Scholar]
- Lehmann, M.; Rocha, S.; Mangeat, B.; Blanchet, F.; Uji-I, H.; Hofkens, J.; Piguet, V. Quantitative multicolor super-resolution microscopy reveals tetherin HIV-1 interaction. PLoS Pathog. 2011, 7, e1002456. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Meth. 2008, 5, 155–157. [Google Scholar]
- Malkusch, S.; Muranyi, W.; Müller, B.; Kräusslich, H.-G.; Heilemann, M. Single-molecule coordinate-based analysis of the morphology of HIV-1 assembly sites with near-molecular spatial resolution. Histochem. Cell Biol. 2012, 139, 173–179. [Google Scholar]
- Muranyi, W.; Malkusch, S.; Müller, B.; Heilemann, M.; Kräusslich, H.-G. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal Tail. PLoS Pathog. 2013, 9, e1003198. [Google Scholar] [CrossRef]
- Roy, N.H.; Chan, J.; Lambelé, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525. [Google Scholar] [CrossRef]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 2014, 343, 653–656. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Muller, B.; Hell, S.W.; Krausslich, H.-G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef]
- Pereira, C.F.; Rossy, J.; Owen, D.M.; Mak, J.; Gaus, K. HIV taken by STORM: Super-resolution fluorescence microscopy of a viral infection. Virol J. 2012, 9. [Google Scholar] [CrossRef]
- Charrin, S.; le Naour, F.; Silvie, O.; Milhiet, P.-E.; Boucheix, C.; Rubinstein, E. Lateral organization of membrane proteins: Tetraspanins spin their web. Biochem J. 2009, 420, 133–154. [Google Scholar] [CrossRef]
- Barreiro, O.; Zamai, M.; Yáñez-Mó, M.; Tejera, E.; López-Romero, P.; Monk, P.N.; Gratton, E.; Caiolfa, V.R.; Sanchez-Madrid, F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J. Cell Biol. 2008, 183, 527–542. [Google Scholar] [CrossRef]
- Rocha-Perugini, V.; Zamai, M.; Gonzalez-Granado, J.M.; Barreiro, O.; Tejera, E.; Yanez-Mo, M.; Caiolfa, V.R.; Sanchez-Madrid, F. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol. Cell. Biol. 2013, 33, 3644–3658. [Google Scholar] [CrossRef]
- Nydegger, S. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Cell Biol. 2006, 173, 795–807. [Google Scholar] [CrossRef]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef]
- Jolly, C.; Sattentau, Q.J. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 2007, 81, 7873–7884. [Google Scholar] [CrossRef]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef]
- Harris, H.J.; Davis, C.; Mullins, J.G.L.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 2010, 285, 21092–21102. [Google Scholar] [CrossRef]
- Davis, C.; Harris, H.J.; Hu, K.; Drummer, H.E.; McKeating, J.A.; Mullins, J.G.L.; Balfe, P. In silico directed mutagenesis identifies the CD81/claudin-1 hepatitis C virus receptor interface. Cell. Microbiol. 2012, 14, 1892–1903. [Google Scholar] [CrossRef]
- Potel, J.; Rassam, P.; Montpellier, C.; Kaestner, L.; Werkmeister, E.; Tews, B.A.; Couturier, C.; Popescu, C.-I.; Baumert, T.F.; Rubinstein, E.; et al. EWI-2wint promotes CD81 clustering that abrogates Hepatitis C Virus entry. Cell. Microbiol. 2013, 15, 1234–1252. [Google Scholar] [CrossRef]
- Harris, H.J.; Clerte, C.; Farquhar, M.J.; Goodall, M.; Hu, K.; Rassam, P.; Dosset, P.; Wilson, G.K.; Balfe, P.; IJzendoorn, S.C.; et al. Hepatoma polarization limits CD81 and hepatitis C virus dynamics. Cell. Microbiol. 2013, 15, 430–445. [Google Scholar] [CrossRef]
- Krementsov, D.N.; Rassam, P.; Margeat, E.; Roy, N.H.; Schneider-Schaulies, J.; Milhiet, P.-E.; Thali, M. HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 2010, 11, 1401–1414. [Google Scholar] [CrossRef]
- He, J.; Sun, E.; Bujny, M.V.; Kim, D.; Davidson, M.W.; Zhuang, X. Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathog. 2013, 9, e1003701. [Google Scholar] [CrossRef]
- Mattila, P.K.; Feest, C.; Depoil, D.; Treanor, B.; Montaner, B.; Otipoby, K.L.; Carter, R.; Justement, L.B.; Bruckbauer, A.; Batista, F.D. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity 2013, 38, 461–474. [Google Scholar] [CrossRef]
- Zhang, J.; Leiderman, K.; Pfeiffer, J.R.; Wilson, B.S.; Oliver, J.M.; Steinberg, S.L. Characterizing the topography of membrane receptors and signaling molecules from spatial patterns obtained using nanometer-scale electron-dense probes and electron microscopy. Micron 2006, 37, 14–34. [Google Scholar] [CrossRef]
- Kiskowski, M.A.; Hancock, J.F.; Kenworthy, A.K. On the use of Ripley’s K-function and its derivatives to analyze domain size. Biophys. J. 2009, 97, 1095–1103. [Google Scholar] [CrossRef]
- Sengupta, P.; Jovanovic-Talisman, T.; Skoko, D.; Renz, M.; Veatch, S.L.; Lippincott-Schwartz, J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Meth. 2011, 8, 969–975. [Google Scholar] [CrossRef]
- Rees, E.J.; Erdelyi, M.; Schierle, G.; Knight, A. Elements of image processing in localization microscopy. J. Opt. 2013, 15, 094012. [Google Scholar] [CrossRef]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef]
- Perez-Hernandez, D.; Gutierrez-Vázquez, C.; Jorge, I.; López-Martín, S.; Ursa, A.; Sanchez-Madrid, F.; Vázquez, J.; Yáñez-Mó, M. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J. Biol. Chem. 2013, 288, 11649–11661. [Google Scholar] [CrossRef]
- Quast, T.; Eppler, F.; Semmling, V.; Schild, C.; Homsi, Y.; Levy, S.; Lang, T.; Kurts, C.; Kolanus, W. CD81 is essential for the formation of membrane protrusions and regulates Rac1-activation in adhesion-dependent immune cell migration. Blood 2011, 118, 1818–1827. [Google Scholar] [CrossRef]
- Bari, R.; Guo, Q.; Xia, B.; Zhang, Y.H.; Giesert, E.E.; Levy, S.; Zheng, J.J.; Zhang, X.A. Tetraspanins regulate the protrusive activities of cell membrane. Biochem. Biophys. Res. Commun. 2011, 415, 619–626. [Google Scholar] [CrossRef]
- Holm, T.; Klein, T.; Löschberger, A.; Klamp, T.; Wiebusch, G.; van de Linde, S.; Sauer, M. A Blueprint for cost-efficient localization microscopy. Chemphyschem 2013. [Google Scholar] [CrossRef]
- Henriques, R.; Lelek, M.; Fornasiero, E.F.; Valtorta, F.; Zimmer, C.; Mhlanga, M.M. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Meth. 2010, 7, 339–340. [Google Scholar] [CrossRef]
- Holden, S.J.; Uphoff, S.; Kapanidis, A.N. DAOSTORM: An algorithm for high-density super-resolution microscopy. Nat. Meth. 2011, 8, 279–280. [Google Scholar] [CrossRef]
- Wolter, S.; Löschberger, A.; Holm, T.; Aufmkolk, S.; Dabauvalle, M.-C.; van de Linde, S.; Sauer, M. rapidSTORM: Accurate, fast open-source software for localization microscopy. Nat. Meth. 2012, 9, 1040–1041. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grove, J. Super-Resolution Microscopy: A Virus’ Eye View of the Cell. Viruses 2014, 6, 1365-1378. https://doi.org/10.3390/v6031365
Grove J. Super-Resolution Microscopy: A Virus’ Eye View of the Cell. Viruses. 2014; 6(3):1365-1378. https://doi.org/10.3390/v6031365
Chicago/Turabian StyleGrove, Joe. 2014. "Super-Resolution Microscopy: A Virus’ Eye View of the Cell" Viruses 6, no. 3: 1365-1378. https://doi.org/10.3390/v6031365
APA StyleGrove, J. (2014). Super-Resolution Microscopy: A Virus’ Eye View of the Cell. Viruses, 6(3), 1365-1378. https://doi.org/10.3390/v6031365