Ready, Set, Fuse! The Coronavirus Spike Protein and Acquisition of Fusion Competence

Abstract
:1. Introduction
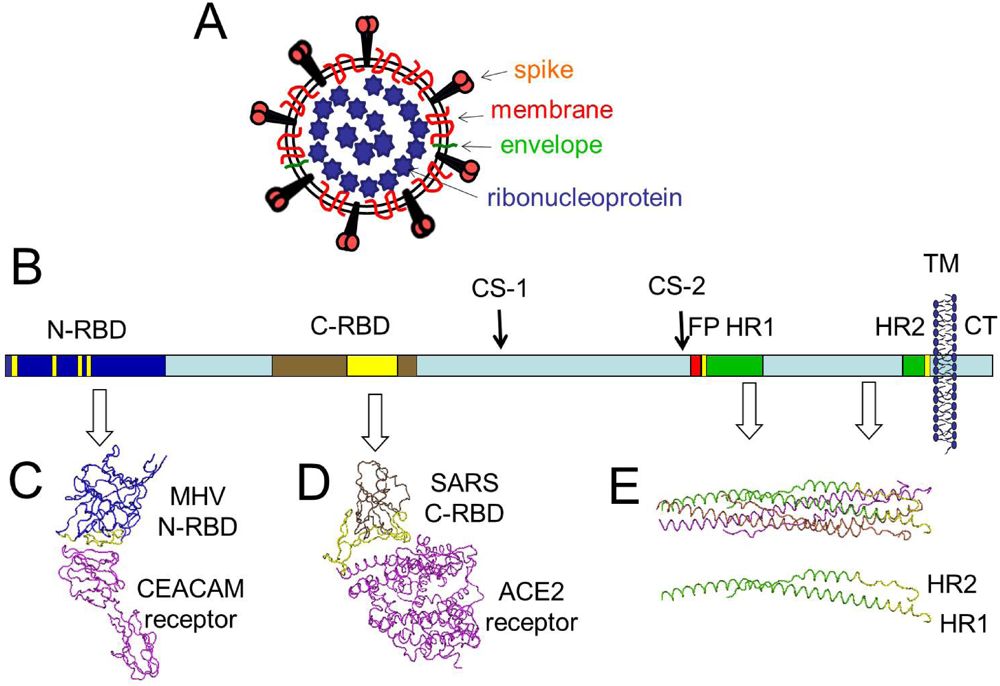
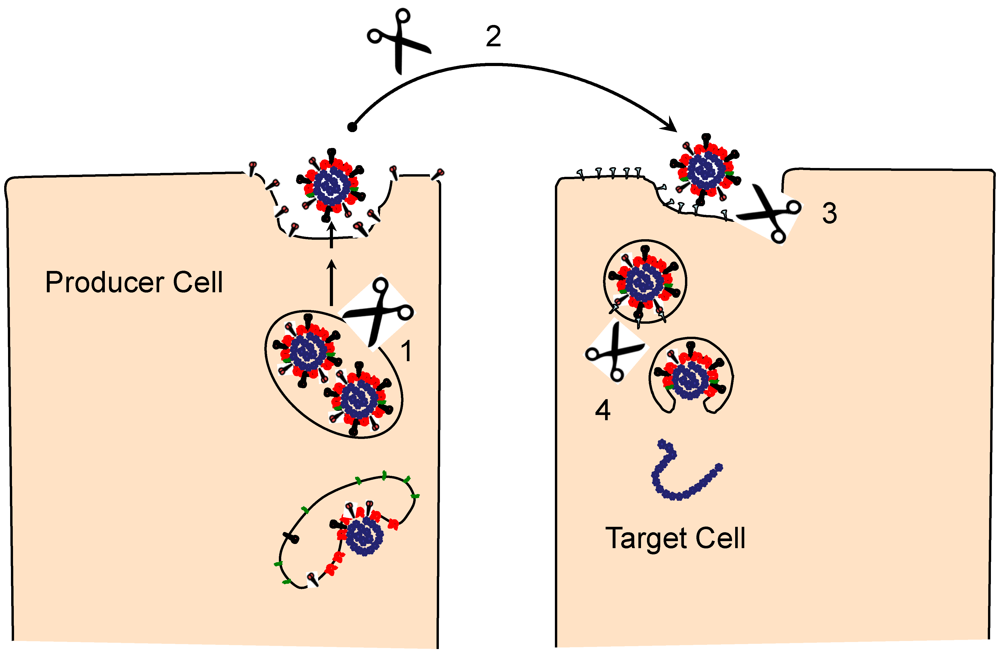
2. S protein Morphogenesis and Initial S Protein Proteolysis
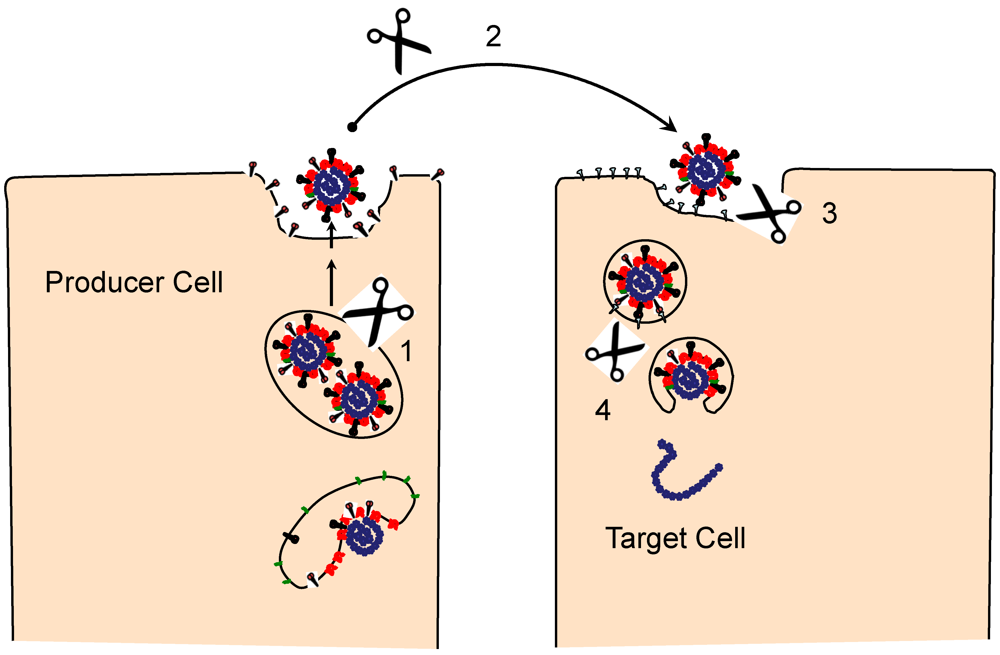
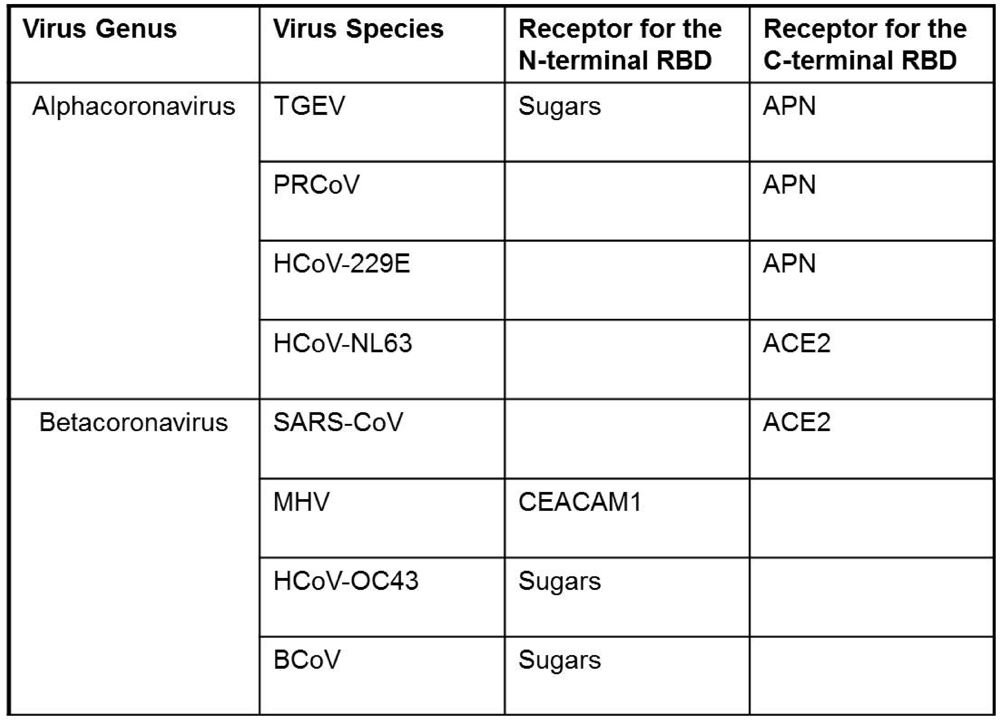
3. Engaging the CoV Receptors

{kind=link}
{kind=link}
{kind=link}
 |
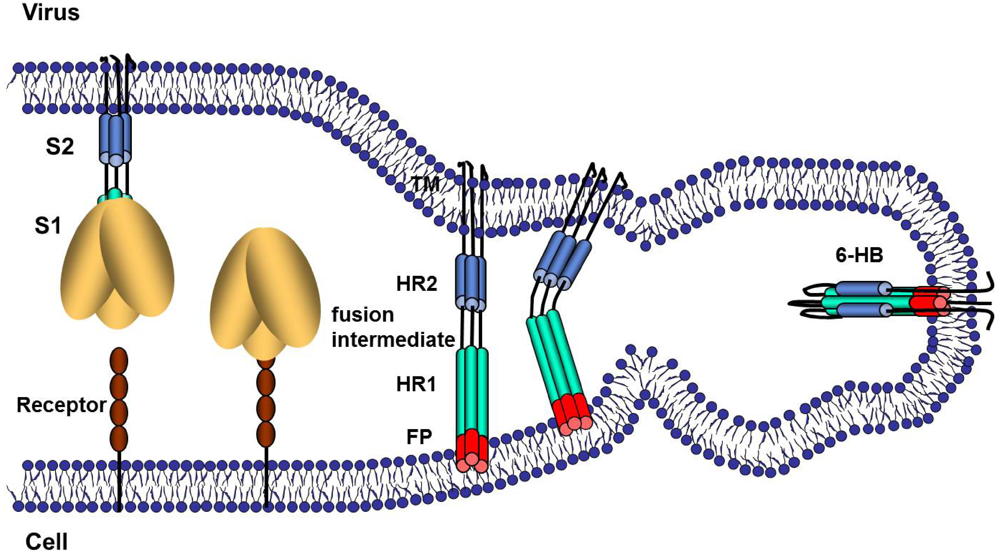
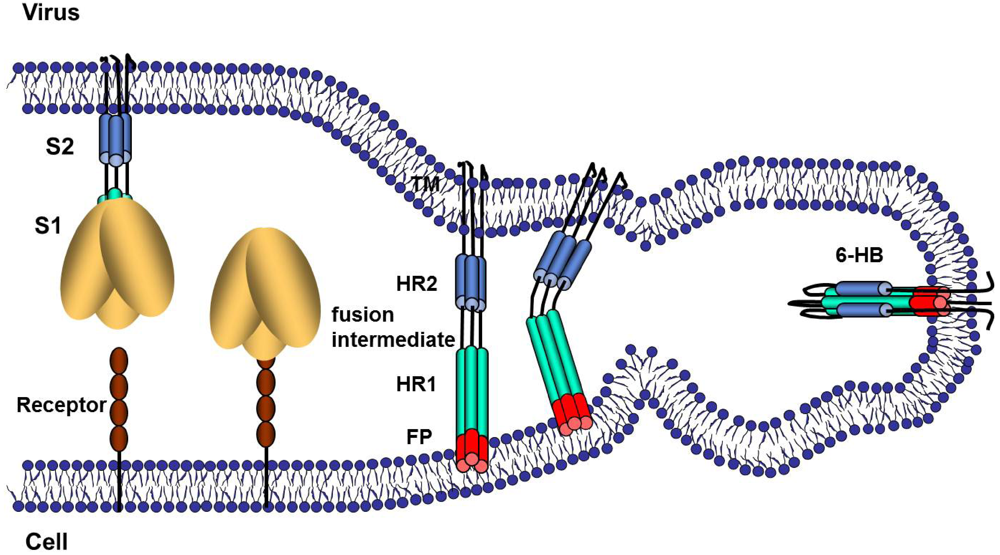
5. Virus Entry, S Proteolysis and Membrane Fusion
6. Therapeutics and Inhibition of CoV Entry
7. Future Directions
Conflict of Interest
References
- Opstelten, D.J.; de Groote, P.; Horzinek, M.C.; Vennema, H.; Rottier, P.J. Disulfide bonds in folding and transport of mouse hepatitis coronavirus glycoproteins. J. Virol. 1993, 67, 7394–7401. [Google Scholar]
- Tooze, J.; Tooze, S.; Warren, G. Replication of coronavirus mhv-a59 in sac- cells: Determination of the first site of budding of progeny virions. Eur. J. Cell Biol. 1984, 33, 281–293. [Google Scholar]
- Neuman, B.W.; Adair, B.D.; Yoshioka, C.; Quispe, J.D.; Milligan, R.A.; Yeager, M.; Buchmeier, M.J. Ultrastructure of sars-cov, fipv, and mhv revealed by electron cryomicroscopy. Adv. Exp. Med. Biol. 2006, 581, 181–185. [Google Scholar]
- Neuman, B.W.; Adair, B.D.; Yoshioka, C.; Quispe, J.D.; Orca, G.; Kuhn, P.; Milligan, R.A.; Yeager, M.; Buchmeier, M.J. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 2006, 80, 7918–7928. [Google Scholar]
- Beniac, D.R.; Andonov, A.; Grudeski, E.; Booth, T.F. Architecture of the sars coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006, 13, 751–752. [Google Scholar]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of nl63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of sars coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar]
- Peng, G.; Sun, D.; Rajashankar, K.R.; Qian, Z.; Holmes, K.V.; Li, F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 10696–10701. [Google Scholar]
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by sars coronavirus. J. Biol. Chem. 2012, 287, 8904–8911. [Google Scholar]
- Xu, Y.; Zhu, J.; Liu, Y.; Lou, Z.; Yuan, F.; Liu, Y.; Cole, D.K.; Ni, L.; Su, N.; Qin, L.; et al. haracterization of the heptad repeat regions, hr1 and hr2, and design of a fusion core structure model of the spike protein from severe acute respiratory syndrome (sars) coronavirus. Biochemistry 2004, 43, 14064–14071. [Google Scholar]
- Deng, Y.; Liu, J.; Zheng, Q.; Yong, W.; Lu, M. Structures and polymorphic interactions of two heptad-repeat regions of the sars virus s2 protein. Structure 2006, 14, 889–899. [Google Scholar]
- Xu, Y.; Liu, Y.; Lou, Z.; Qin, L.; Li, X.; Bai, Z.; Pang, H.; Tien, P.; Gao, G.F.; Rao, Z. Structural basis for coronavirus-mediated membrane fusion. Crystal structure of mouse hepatitis virus spike protein fusion core. J. Biol. Chem. 2004, 279, 30514–30522. [Google Scholar]
- Zheng, Q.; Deng, Y.; Liu, J.; van der Hoek, L.; Berkhout, B.; Lu, M. Core structure of s2 from the human coronavirus nl63 spike glycoprotein. Biochemistry 2006, 45, 15205–15215. [Google Scholar]
- Wu, K.; Chen, L.; Peng, G.; Zhou, W.; Pennell, C.A.; Mansky, L.M.; Geraghty, R.J.; Li, F. A virus-binding hot spot on human angiotensin-converting enzyme 2 is critical for binding of two different coronaviruses. J. Virol. 2011, 85, 5331–5337. [Google Scholar]
- Duquerroy, S.; Vigouroux, A.; Rottier, P.J.; Rey, F.A.; Bosch, B.J. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the sars coronavirus spike glycoprotein. Virology 2005, 335, 276–285. [Google Scholar]
- Lamb, R.A.; Jardetzky, T.S. Structural basis of viral invasion: Lessons from paramyxovirus f. Curr. Opin. Struct. Biol. 2007, 17, 427–436. [Google Scholar]
- Luo, Z.; Weiss, S.R. Roles in cell-to-cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology 1998, 244, 483–494. [Google Scholar]
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein s2 domain with characteristics of a viral fusion peptide. J. Virol. 2009, 83, 7411–7421. [Google Scholar]
- Broer, R.; Boson, B.; Spaan, W.; Cosset, F.L.; Corver, J. Important role for the transmembrane domain of severe acute respiratory syndrome coronavirus spike protein during entry. J. Virol. 2006, 80, 1302–1310. [Google Scholar]
- Lu, Y.; Neo, T.L.; Liu, D.X.; Tam, J.P. Importance of sars-cov spike protein trp-rich region in viral infectivity. Biochem. Biophys. Res. Commun. 2008, 371, 356–360. [Google Scholar]
- Bos, E.C.; Heijnen, L.; Luytjes, W.; Spaan, W.J. Mutational analysis of the murine coronavirus spike protein: Effect on cell-to-cell fusion. Virology 1995, 214, 453–463. [Google Scholar]
- Petit, C.M.; Chouljenko, V.N.; Iyer, A.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Palmitoylation of the cysteine-rich endodomain of the sars-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. [Google Scholar]
- Shulla, A.; Gallagher, T. Role of spike protein endodomains in regulating coronavirus entry. J. Biol. Chem. 2009, 284, 32725–32734. [Google Scholar]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class i virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar]
- Tripet, B.; Howard, M.W.; Jobling, M.; Holmes, R.K.; Holmes, K.V.; Hodges, R.S. Structural characterization of the sars-coronavirus spike s fusion protein core. J. Biol. Chem. 2004, 279, 20836–20849. [Google Scholar]
- Bosch, B.J.; Martina, B.E.; Van Der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.; De Groot, R.; Osterhaus, A.D.; Rottier, P.J. Severe acute respiratory syndrome coronavirus (sars-cov) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natil. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar]
- Liu, I.J.; Kao, C.L.; Hsieh, S.C.; Wey, M.T.; Kan, L.S.; Wang, W.K. Identification of a minimal peptide derived from heptad repeat (hr) 2 of spike protein of sars-cov and combination of hr1-derived peptides as fusion inhibitors. Antiviral Res. 2009, 81, 82–87. [Google Scholar]
- Zheng, B.J.; Guan, Y.; Hez, M.L.; Sun, H.; Du, L.; Zheng, Y.; Wong, K.L.; Chen, H.; Chen, Y.; Lu, L.; et al. Synthetic peptides outside the spike protein heptad repeat regions as potent inhibitors of sars-associated coronavirus. Antiviral Ther. 2005, 10, 393–403. [Google Scholar]
- Sturman, L.S.; Ricard, C.S.; Holmes, K.V. Conformational change of the coronavirus peplomer glycoprotein at ph 8.0 and 37 degrees c correlates with virus aggregation and virus-induced cell fusion. J. Virol. 1990, 64, 3042–3050. [Google Scholar]
- Gallagher, T.M. A role for naturally occurring variation of the murine coronavirus spike protein in stabilizing association with the cellular receptor. J. Virol. 1997, 71, 3129–3137. [Google Scholar]
- Ruch, T.R.; Machamer, C.E. The hydrophobic domain of infectious bronchitis virus e protein alters the host secretory pathway and is important for release of infectious virus. J. Virol. 2011, 85, 675–685. [Google Scholar]
- Nieto-Torres, J.L.; Dediego, M.L.; Alvarez, E.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. Sars coronavirus e protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar]
- Ortego, J.; Ceriani, J.E.; Patino, C.; Plana, J.; Enjuanes, L. Absence of e protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308. [Google Scholar]
- Feliciangeli, S.F.; Thomas, L.; Scott, G.K.; Subbian, E.; Hung, C.H.; Molloy, S.S.; Jean, F.; Shinde, U.; Thomas, G. Identification of a ph sensor in the furin propeptide that regulates enzyme activation. J. Biol. Chem. 2006, 281, 16108–16116. [Google Scholar]
- de Haan, C.A.; Stadler, K.; Godeke, G.J.; Bosch, B.J.; Rottier, P.J. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J. Virol. 2004, 78, 6048–6054. [Google Scholar]
- Gombold, J.L.; Hingley, S.T.; Weiss, S.R. Fusion-defective mutants of mouse hepatitis virus a59 contain a mutation in the spike protein cleavage signal. J. Virol. 1993, 67, 4504–4512. [Google Scholar]
- Yamada, Y.K.; Takimoto, K.; Yabe, M.; Taguchi, F. Acquired fusion activity of a murine coronavirus mhv-2 variant with mutations in the proteolytic cleavage site and the signal sequence of the s protein. Virology 1997, 227, 215–219. [Google Scholar]
- Frana, M.F.; Behnke, J.N.; Sturman, L.S.; Holmes, K.V. Proteolytic cleavage of the e2 glycoprotein of murine coronavirus: Host-dependent differences in proteolytic cleavage and cell fusion. J. Virol. 1985, 56, 912–920. [Google Scholar]
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the sars coronavirus spike glycoprotein enhances cell-cell fusion but does not affect virion entry. Virology 2006, 350, 358–369. [Google Scholar]
- de Haan, C.A.; Li, Z.; te Lintelo, E.; Bosch, B.J.; Haijema, B.J.; Rottier, P.J. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J. Virol. 2005, 79, 14451–14456. [Google Scholar]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ace2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar]
- Thorp, E.B.; Gallagher, T.M. Requirements for ceacams and cholesterol during murine coronavirus cell entry. J. Virol. 2004, 78, 2682–2692. [Google Scholar]
- Watanabe, R.; Matsuyama, S.; Taguchi, F. Receptor-independent infection of murine coronavirus: Analysis by spinoculation. J. Virol. 2006, 80, 4901–4908. [Google Scholar]
- Miura, H.S.; Nakagaki, K.; Taguchi, F. N-terminal domain of the murine coronavirus receptor ceacam1 is responsible for fusogenic activation and conformational changes of the spike protein. J. Virol. 2004, 78, 216–223. [Google Scholar]
- Matsuyama, S.; Taguchi, F. Receptor-induced conformational changes of murine coronavirus spike protein. J. Virol. 2002, 76, 11819–11826. [Google Scholar]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of sars-coronavirus adaptation to human ace2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef]
- Becker, M.M.; Graham, R.L.; Donaldson, E.F.; Rockx, B.; Sims, A.C.; Sheahan, T.; Pickles, R.J.; Corti, D.; Johnston, R.E.; Baric, R.S.; et al. Synthetic recombinant bat sars-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19944–19949. [Google Scholar]
- Gallagher, T.M.; Parker, S.E.; Buchmeier, M.J. Neutralization-resistant variants of a neurotropic coronavirus are generated by deletions within the amino-terminal half of the spike glycoprotein. J. Virol. 1990, 64, 731–741. [Google Scholar]
- Rowe, C.L.; Fleming, J.O.; Nathan, M.J.; Sgro, J.Y.; Palmenberg, A.C.; Baker, S.C. Generation of coronavirus spike deletion variants by high-frequency recombination at regions of predicted rna secondary structure. J. Virol. 1997, 71, 6183–6190. [Google Scholar]
- Phillips, J.M.; Weiss, S.R. Pathogenesis of neurotropic murine coronavirus is multifactorial. Trends Pharmacol. Sci. 2011, 32, 2–7. [Google Scholar]
- Enjuanes, L.; Sanchez, C.; Gebauer, F.; Mendez, A.; Dopazo, J.; Ballesteros, M.L. Evolution and tropism of transmissible gastroenteritis coronavirus. Adv. Exp. Med. Biol. 1993, 342, 35–42. [Google Scholar]
- Schultze, B.; Krempl, C.; Ballesteros, M.L.; Shaw, L.; Schauer, R.; Enjuanes, L.; Herrler, G. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (n-glycolylneuraminic acid) binding activity. J. Virol. 1996, 70, 5634–5637. [Google Scholar]
- Sanchez, C.M.; Gebauer, F.; Sune, C.; Mendez, A.; Dopazo, J.; Enjuanes, L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 1992, 190, 92–105. [Google Scholar]
- Ballesteros, M.L.; Sanchez, C.M.; Enjuanes, L. Two amino acid changes at the n-terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology 1997, 227, 378–388. [Google Scholar]
- Zelus, B.D.; Schickli, J.H.; Blau, D.M.; Weiss, S.R.; Holmes, K.V. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees c either by soluble murine ceacam1 receptors or by ph 8. J. Virol. 2003, 77, 830–840. [Google Scholar]
- Matsuyama, S.; Taguchi, F. Two-step conformational changes in a coronavirus envelope glycoprotein mediated by receptor binding and proteolysis. J. Virol. 2009, 83, 11133–11141. [Google Scholar]
- Grosse, B.; Siddell, S.G. Single amino acid changes in the s2 subunit of the mhv surface glycoprotein confer resistance to neutralization by s1 subunit-specific monoclonal antibody. Virology 1994, 202, 814–824. [Google Scholar]
- Ontiveros, E.; Kim, T.S.; Gallagher, T.M.; Perlman, S. Enhanced virulence mediated by the murine coronavirus, mouse hepatitis virus strain jhm, is associated with a glycine at residue 310 of the spike glycoprotein. J. Virol. 2003, 77, 10260–10269. [Google Scholar]
- de Haan, C.A.; Te Lintelo, E.; Li, Z.; Raaben, M.; Wurdinger, T.; Bosch, B.J.; Rottier, P.J. Cooperative involvement of the s1 and s2 subunits of the murine coronavirus spike protein in receptor binding and extended host range. J. Virol. 2006, 80, 10909–10918. [Google Scholar]
- Matsuyama, S.; Taguchi, F. Communication between s1n330 and a region in s2 of murine coronavirus spike protein is important for virus entry into cells expressing ceacam1b receptor. Virology 2002, 295, 160–171. [Google Scholar]
- Krueger, D.K.; Kelly, S.M.; Lewicki, D.N.; Ruffolo, R.; Gallagher, T.M. Variations in disparate regions of the murine coronavirus spike protein impact the initiation of membrane fusion. J. Virol. 2001, 75, 2792–2802. [Google Scholar]
- Sieczkarski, S.B.; Whittaker, G.R. Viral entry. Curr. Top. Microbiol. Immunol. 2005, 285, 1–23. [Google Scholar]
- Marsh, M.; Bron, R. Sfv infection in cho cells: Cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 1997, 110 (Pt 1), 95–103. [Google Scholar]
- Gallagher, T.M.; Escarmis, C.; Buchmeier, M.J. Alteration of the ph dependence of coronavirus-induced cell fusion: Effect of mutations in the spike glycoprotein. J. Virol. 1991, 65, 1916–1928. [Google Scholar]
- Roos, D.S.; Duchala, C.S.; Stephensen, C.B.; Holmes, K.V.; Choppin, P.W. Control of virus-induced cell fusion by host cell lipid composition. Virology 1990, 175, 345–357. [Google Scholar]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. Hiv enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar]
- Sieczkarski, S.B.; Whittaker, G.R. Dissecting virus entry via endocytosis. J. Gen. Virol. 2002, 83, 1535–1545. [Google Scholar]
- Nomura, R.; Kiyota, A.; Suzaki, E.; Kataoka, K.; Ohe, Y.; Miyamoto, K.; Senda, T.; Fujimoto, T. Human coronavirus 229e binds to cd13 in rafts and enters the cell through caveolae. J. Virol. 2004, 78, 8701–8708. [Google Scholar]
- Choi, K.S.; Aizaki, H.; Lai, M.M. Murine coronavirus requires lipid rafts for virus entry and cell-cell fusion but not for virus release. J. Virol. 2005, 79, 9862–9871. [Google Scholar]
- Van Hamme, E.; Dewerchin, H.L.; Cornelissen, E.; Verhasselt, B.; Nauwynck, H.J. Clathrin- and caveolae-independent entry of feline infectious peritonitis virus in monocytes depends on dynamin. J. Gen. Virol. 2008, 89, 2147–2156. [Google Scholar]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. Sars coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar]
- Jaume, M.; Yip, M.S.; Cheung, C.Y.; Leung, H.L.; Li, P.H.; Kien, F.; Dutry, I.; Callendret, B.; Escriou, N.; Altmeyer, R.; et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a ph- and cysteine protease-independent fcgammar pathway. J. Virol. 2011, 85, 10582–10597. [Google Scholar]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (sars-cov) spike glycoprotein-mediated viral entry. Proc. Natil. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar]
- Qiu, Z.; Hingley, S.T.; Simmons, G.; Yu, C.; Das Sarma, J.; Bates, P.; Weiss, S.R. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006, 80, 5768–5776. [Google Scholar]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin l prevent severe acute respiratory syndrome coronavirus entry. Proc. Natil. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar]
- Gregory, S.M.; Harada, E.; Liang, B.; Delos, S.E.; White, J.M.; Tamm, L.K. Structure and function of the complete internal fusion loop from ebolavirus glycoprotein 2. Proc. Natil. Acad. Sci. USA 2011, 108, 11211–11216. [Google Scholar]
- McReynolds, S.; Jiang, S.; Guo, Y.; Celigoy, J.; Schar, C.; Rong, L.; Caffrey, M. Characterization of the prefusion and transition states of severe acute respiratory syndrome coronavirus s2-hr2. Biochemistry 2008, 47, 6802–6808. [Google Scholar]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natil. Acad. Sci. USA 2005, 102, 12543–12547. [Google Scholar]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J. Cathepsin l functionally cleaves the severe acute respiratory syndrome coronavirus class i fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved s protein as revealed by pseudotype virus bearing cleaved s protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar]
- Yamada, Y.; Liu, D.X. Proteolytic activation of the spike protein at a novel rrrr/s motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J. Virol. 2009, 83, 8744–8758. [Google Scholar]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the sars coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natil. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar]
- Kam, Y.W.; Okumura, Y.; Kido, H.; Ng, L.F.; Bruzzone, R.; Altmeyer, R. Cleavage of the sars coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One 2009, 4, e7870. [Google Scholar]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease tmprss2. J. Virol. 2010, 84, 12658–12664. [Google Scholar]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that tmprss2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar]
- Bertram, S.; Glowacka, I.; Muller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; Soilleux, E.J.; Jahn, O.; Steffen, I.; Pohlmann, S. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar]
- Szabo, R.; Bugge, T.H. Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu. Rev. Cell Dev. Biol. 2011, 27, 213–235. [Google Scholar]
- Hooper, J.D.; Clements, J.A.; Quigley, J.P.; Antalis, T.M. Type ii transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J. Biol. Chem. 2001, 276, 857–860. [Google Scholar]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the tmprss2 gene, which encodes a novel serine protease with transmembrane, ldlra, and srcr domains and maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic analysis of mice lacking the tmprss2-encoded protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases tmprss2 and hat from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Tsegaye, T.S.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; Marzi, A.; Pohlmann, S. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 2009, 83, 3200–3211. [Google Scholar]
- Bottcher-Friebertshauser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of influenza virus hemagglutinin by airway proteases tmprss2 and hat differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar]
- Okumura, Y.; Takahashi, E.; Yano, M.; Ohuchi, M.; Daidoji, T.; Nakaya, T.; Bottcher, E.; Garten, W.; Klenk, H.D.; Kido, H. Novel type ii transmembrane serine proteases, mspl and tmprss13, proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J. Virol. 2010, 84, 5089–5096. [Google Scholar]
- Huang, I.C.; Bosch, B.J.; Li, F.; Li, W.; Lee, K.H.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. Sars coronavirus, but not human coronavirus nl63, utilizes cathepsin l to infect ace2-expressing cells. J. Biol. Chem. 2006, 281, 3198–3203. [Google Scholar]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of sars-cov--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar]
- van den Brink, E.N.; Ter Meulen, J.; Cox, F.; Jongeneelen, M.A.; Thijsse, A.; Throsby, M.; Marissen, W.E.; Rood, P.M.; Bakker, A.B.; Gelderblom, H.R.; et al. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1635–1644. [Google Scholar]
- Chen, Z.; Zhang, L.; Qin, C.; Ba, L.; Yi, C.E.; Zhang, F.; Wei, Q.; He, T.; Yu, W.; Yu, J.; et al. Recombinant modified vaccinia virus ankara expressing the spike glycoprotein of severe acute respiratory syndrome coronavirus induces protective neutralizing antibodies primarily targeting the receptor binding region. J. Virol. 2005, 79, 2678–2688. [Google Scholar]
- Miyoshi-Akiyama, T.; Ishida, I.; Fukushi, M.; Yamaguchi, K.; Matsuoka, Y.; Ishihara, T.; Tsukahara, M.; Hatakeyama, S.; Itoh, N.; Morisawa, A.; et al. Fully human monoclonal antibody directed to proteolytic cleavage site in severe acute respiratory syndrome (sars) coronavirus s protein neutralizes the virus in a rhesus macaque sars model. J. Infect. Dis. 2011, 203, 1574–1581. [Google Scholar]
- Mori, T.; O'Keefe, B.R.; Sowder, R.C., 2nd; Bringans, S.; Gardella, R.; Berg, S.; Cochran, P.; Turpin, J.A.; Buckheit, R.W., Jr.; McMahon, J.B.; Boyd, M.R. Isolation and characterization of griffithsin, a novel hiv-inactivating protein, from the red alga griffithsia sp. J. Biol. Chem. 2005, 280, 9345–9353. [Google Scholar]
- O'Keefe, B.R.; Giomarelli, B.; Barnard, D.L.; Shenoy, S.R.; Chan, P.K.; McMahon, J.B.; Palmer, K.E.; Barnett, B.W.; Meyerholz, D.K.; Wohlford-Lenane, C.L.; et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family coronaviridae. J. Virol. 2010, 84, 2511–2521. [Google Scholar]
- Zhou, Y.; Lu, K.; Pfefferle, S.; Bertram, S.; Glowacka, I.; Drosten, C.; Pohlmann, S.; Simmons, G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 2010, 84, 8753–8764. [Google Scholar]
- van der Meer, F.J.; de Haan, C.A.; Schuurman, N.M.; Haijema, B.J.; Peumans, W.J.; van Damme, E.J.; Delputte, P.L.; Balzarini, J.; Egberink, H.F. Antiviral activity of carbohydrate-binding agents against nidovirales in cell culture. Antiviral Res. 2007, 76, 21–29. [Google Scholar]
- Shah, P.P.; Wang, T.; Kaletsky, R.L.; Myers, M.C.; Purvis, J.E.; Jing, H.; Huryn, D.M.; Greenbaum, D.C.; Smith, A.B., 3rd; Bates, P.; et al. A small-molecule oxocarbazate inhibitor of human cathepsin l blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293t cells. Mol. Pharmacol. 2010, 78, 319–324. [Google Scholar]
- Bahgat, M.M.; Blazejewska, P.; Schughart, K. Inhibition of lung serine proteases in mice: A potentially new approach to control influenza infection. Virol. J. 2011, 8, 27. [Google Scholar]
- Lee, M.G.; Kim, K.H.; Park, K.Y.; Kim, J.S. Evaluation of anti-influenza effects of camostat in mice infected with non-adapted human influenza viruses. Arch. Virol. 1996, 141, 1979–1989. [Google Scholar]
- Zhirnov, O.P.; Kirzhner, L.S.; Ovcharenko, A.V.; Malyshev, N.A. clinical effectiveness of aprotinin aerosol in influenza and parainfluenza. Vestnik Rossiiskoi Akademii Meditsinskikh Nauk / Rossiiskaia Akademiia Meditsinskikh Nauk 1996, 26–31. [Google Scholar]
- Bottcher-Friebertshauser, E.; Stein, D.A.; Klenk, H.D.; Garten, W. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin-activating protease tmprss2. J. Virol. 2011, 85, 1554–1562. [Google Scholar]
- Zhu, J.; Xiao, G.; Xu, Y.; Yuan, F.; Zheng, C.; Liu, Y.; Yan, H.; Cole, D.K.; Bell, J.I.; Rao, Z.; et al. Following the rule: Formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 2004, 319, 283–288. [Google Scholar] [CrossRef]
- Tremblay, C.L.; Kollmann, C.; Giguel, F.; Chou, T.C.; Hirsch, M.S. Strong in vitro synergy between the fusion inhibitor t-20 and the cxcr4 blocker amd-3100. J. Acquir Immune Defic. Syndr. 2000, 25, 99–102. [Google Scholar]
- Bosch, B.J.; Rossen, J.W.; Bartelink, W.; Zuurveen, S.J.; de Haan, C.A.; Duquerroy, S.; Boucher, C.A.; Rottier, P.J. Coronavirus escape from heptad repeat 2 (hr2)-derived peptide entry inhibition as a result of mutations in the hr1 domain of the spike fusion protein. J. Virol. 2008, 82, 2580–2585. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heald-Sargent, T.; Gallagher, T. Ready, Set, Fuse! The Coronavirus Spike Protein and Acquisition of Fusion Competence. Viruses 2012, 4, 557-580. https://doi.org/10.3390/v4040557
Heald-Sargent T, Gallagher T. Ready, Set, Fuse! The Coronavirus Spike Protein and Acquisition of Fusion Competence. Viruses. 2012; 4(4):557-580. https://doi.org/10.3390/v4040557
Chicago/Turabian StyleHeald-Sargent, Taylor, and Tom Gallagher. 2012. "Ready, Set, Fuse! The Coronavirus Spike Protein and Acquisition of Fusion Competence" Viruses 4, no. 4: 557-580. https://doi.org/10.3390/v4040557
APA StyleHeald-Sargent, T., & Gallagher, T. (2012). Ready, Set, Fuse! The Coronavirus Spike Protein and Acquisition of Fusion Competence. Viruses, 4(4), 557-580. https://doi.org/10.3390/v4040557