RNA-Sequencing Analysis of 5' Capped RNAs Identifies Many New Differentially Expressed Genes in Acute Hepatitis C Virus Infection

Abstract
:1. Introduction
2. Results
2.1. HCV JFH-1 Infection of Huh 7.5 Cells
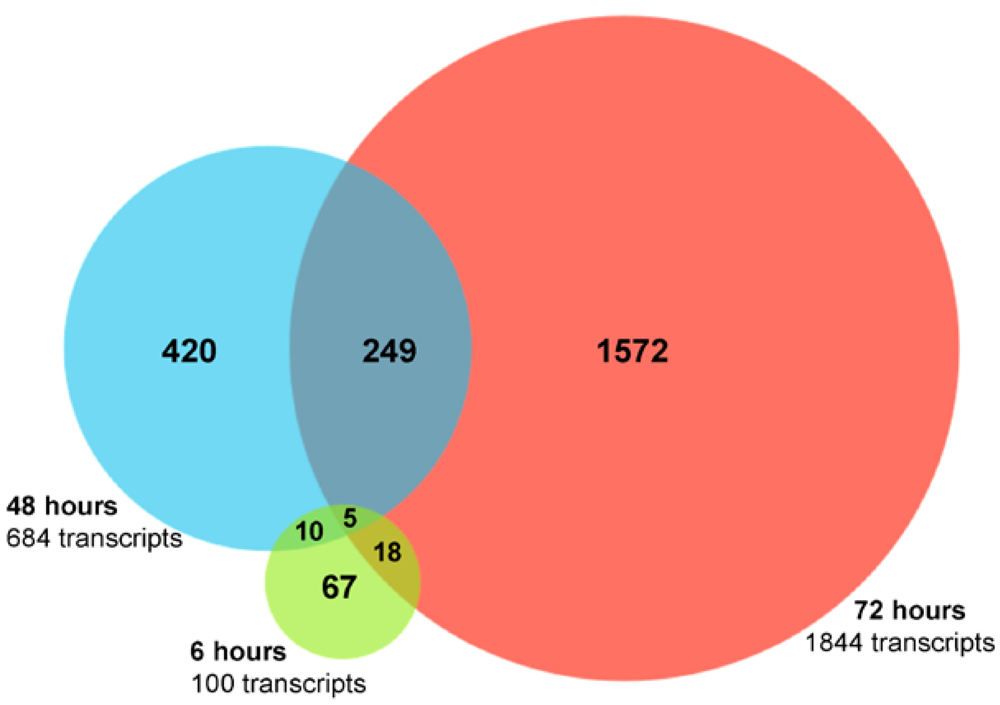
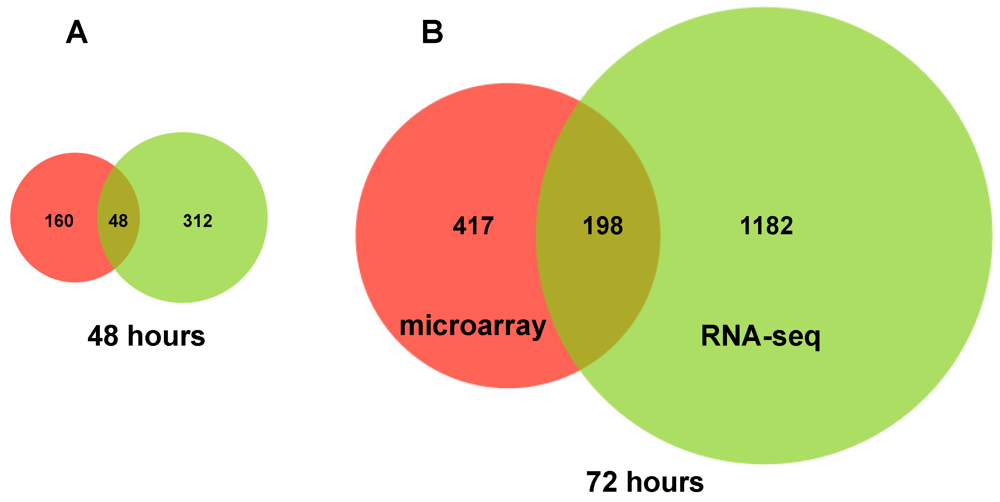
2.2. RNA Sequencing (RNA-seq) Analysis of Annotated Gene Transcripts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl Gene ID | Gene Name | Description | Fold Change | FDR |
|---|---|---|---|---|
| ENSG00000174951 | FUT1 | Fucosyltransferase 1 | 20.51 | 104.09 |
| ENSG00000130487 | KLHDC7B | Kelch domain containing 7 | 18.93 | 141.94 |
| ENSG00000136826 | KLF4 | Kruppel-like factor 4 | 15.10 | 142.63 |
| ENSG00000128591 | FLNC | Filamin C | 13.38 | 1160.20 |
| ENSG00000148677 | ANKRD1 | Ankyrin repeat domain 1 | 12.06 | 351.26 |
| ENSG00000205595 | AREG | Amphiregulin | 11.63 | 229.81 |
| ENSG00000135842 | C1orf24 | Chromosome 1 open reading frame 24 (C1orf24) | 7.23 | 63.37 |
| ENSG00000180535 | MIST1 | Xlass II bHLH protein MIST1 | 6.99 | 84.43 |
| ENSG00000136383 | ALPK3 | Alpha-kinase 3 | 6.83 | 202.42 |
| ENSG00000139269 | INHBE | Inhibin, beta E | 6.40 | 1324.33 |
| ENSG00000107731 | UNC5B | Unc-5 homolog B (C. elegans) | 6.17 | 265.31 |
| ENSG00000049323 | LTBP1 | Latent transforming growth factor beta binding protein 1 | 6.14 | 129.49 |
| ENSG00000048052 | HDAC9 | Histone deacetylase 9 | 6.09 | 137.83 |
| ENSG00000198205 | ZXDA | Zinc finger, X-linked, duplicated A | 6.02 | 47.22 |
| ENSG00000111981 | ULBP1 | UL16 binding protein 1 | 6.01 | 37.66 |
| ENSG00000130513 | GDF15 | Growth differentiation factor 15 | 5.90 | 958.30 |
| ENSG00000122728 | TAF1L | TAF1-like RNA polymerase II, TATA box binding protein (TBP)-associated factor | 5.54 | 41.06 |
| ENSG00000138670 | RASGEF1B | RasGEF domain family, member 1B | 5.41 | 39.54 |
| ENSG00000176971 | LOC387758 | Similar to RIKEN cDNA 1110018M03 | 5.39 | 23.55 |
| ENSG00000160999 | APS | Adaptor protein with pleckstrin homology and src homology 2 domains | 5.18 | 22.09 |
| ENSG00000006327 | TNFRSF12A | Fucosyltransferase 1 | 5.13 | 286.27 |
| ENSG00000184545 | DUSP8* | Dual specificity phosphatase 8 | 5.05 | 87.30 |
| ENSG00000130766 | SESN2 | Sestrin 2 | 4.84 | 593.42 |
| ENSG00000070669 | ASNS | Asparagine synthetase | 4.80 | 577.30 |
| ENSG00000136997 | MYC | v-myc myelocytomatosis viral oncogene homolog | 4.71 | 991.67 |
| ENSG00000168679 | SLC16A4 | Solute carrier family 16 (monocarboxylic acid transporters), member 4 | 4.65 | 24.23 |
| ENSG00000099889 | ARVCF | Armadillo repeat gene deletes in velocardiofacial syndrome | 4.53 | 240.65 |
| ENSG00000162772 | ATF3* | Activating transcription factor 3 | 4.47 | 695.61 |
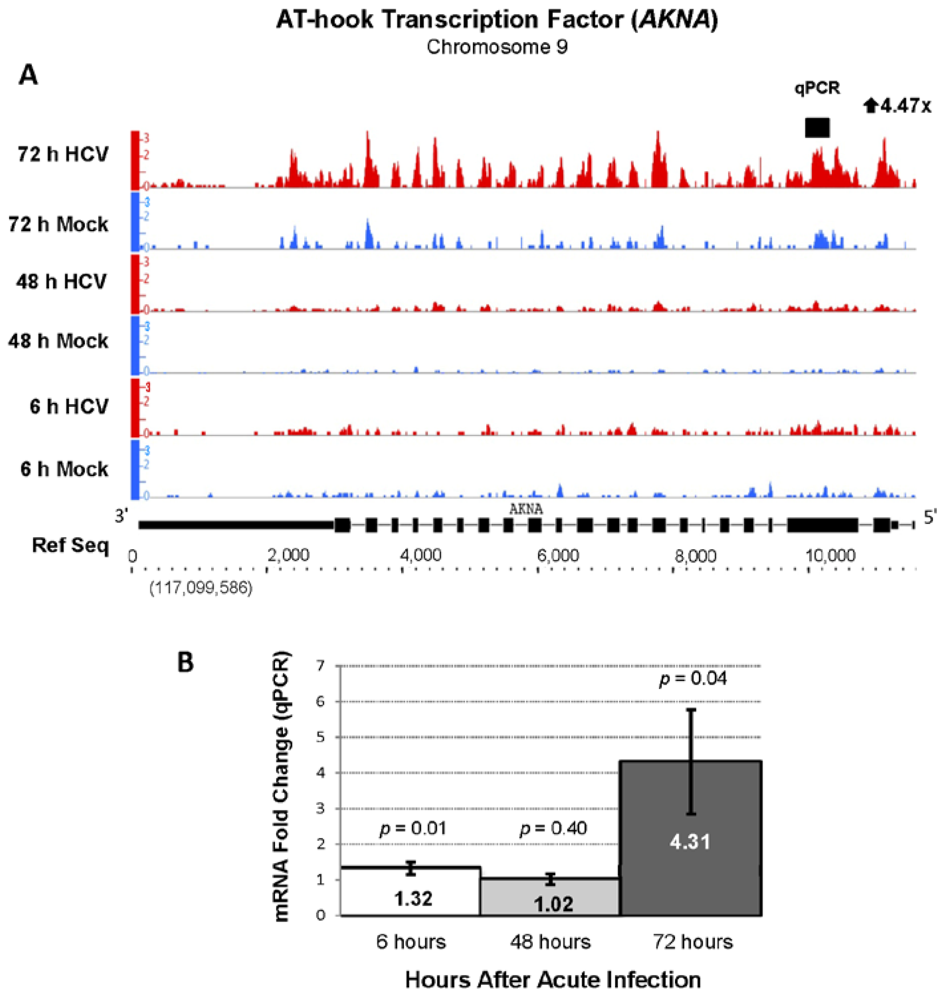
| ENSG00000106948 | AKNA | AT-hook transcription factor | 4.45 | 773.28 |
| ENSG00000167861 | C17orf28 | Chromosome 17 open reading frame 28 | 4.43 | 142.87 |
| ENSG00000138685 | FGF2 | Fibroblast growth factor 2 | 4.08 | 23.78 |
| ENSG00000105327 | BBC3 | BCL2 binding component 3 | 4.06 | 230.60 |
| ENSG00000111087 | GLI1 | Glioma-associated oncogene homolog 1 (zinc finger protein) | 4.02 | 41.66 |
| ENSG00000065911 | MTHFD2 | Methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2, | 4.00 | 693.07 |
| ENSG00000115520 | COQ10B | Coenzyme Q10 homolog B (S. cerevisiae) | 3.96 | 22.43 |
| ENSG00000105499 | PLA2G4C* | Phospholipase A2, group IVC (cytosolic, calcium-independent) | 3.92 | 44.14 |
| ENSG00000065600 | TMEM206 | Transmembrane protein 206 | 3.90 | 57.11 |
| ENSG00000108551 | RASD1 | RAS, dexamethasone-induced 1 | 3.89 | 70.16 |
| ENSG00000152137 | HSPB8 | Heat shock 22kDa protein 8 | 3.87 | 64.97 |
| ENSG00000112182 | BACH2 | BTB and CNC homology 1, basic leucine zipper transcription factor 2 | 3.83 | 76.67 |
| ENSG00000157765 | SLC34A2 | Homo sapiens cDNA FLJ90534 fis, highly similar to Homo sapiens sodium dependent phosphate transporter isoform NaPi-3b mRNA | 3.80 | 41.45 |
| ENSG00000163393 | SLC22A15 | Solute carrier family 22 (organic cation transporter), member 15 | 3.77 | 104.50 |
| ENSG00000168003 | SLC3A2 | Solute carrier family 3 (activators of dibasic and neutral amino acid transport), member 2 | 3.77 | 1977.88 |
| ENSG00000164949 | GEM | GTP binding protein overexpressed in skeletal muscle (GEM) | 3.71 | 51.70 |
| ENSG00000107281 | NPDC1 | Neural proliferation, differentiation and control, 1 | 3.68 | 38.82 |
| ENSG00000154025 | SLC5A10 | Solute carrier family 5 (sodium/glucose cotransporter), member 10 | 3.67 | 50.35 |
| ENSG00000196517 | SLC6A9 | Solute carrier family 6 (neurotransmitter transporter, glycine), member 9 | 3.65 | 153.70 |
| ENSG00000166173 | LARP6 | La ribonuleoprotein domain family, member 6 | 3.62 | 146.96 |
| ENSG00000184371 | CSF1 | Colony stimulating factor 1 (macrophage) | 3.58 | 132.33 |
| ENSG00000120129 | DUSP1 | Dual specificity phosphatase 1 (DUSP1) | 3.53 | 446.22 |
| Ensembl Gene ID | Gene Name | Description | Fold Change | FDR | Time Point |
|---|---|---|---|---|---|
| ENSG00000196475 | GK2 | Glycerol kinase 2 | −17.49 | −22.37 | 72 h |
| ENSG00000164588 | HCN1 | Hyperpolarization activated cyclic nucleotide-gated potassium channel 1 | −15.64 | −23.39 | 6 h |
| ENSG00000101438 | SLC32A1 | Solute carrier family 32 (GABA vesicular transporter), member 1 | −9.34 | −68.18 | 72 h |
| ENSG00000178695 | KCTD12 | Potassium channel tetramerisation domain containing 12 | −8.10 | −41.92 | 6 h |
| ENSG00000182687 | GALR2 | Galanin receptor 2 | −6.23 | −17.14 | 6 h |
| ENSG00000120738 | EGR1 | Early growth response 1 | −5.33 | −259.33 | 6 h |
| ENSG00000170989 | EDG1 | Endothelial differentiation, sphingolipid G-protein-coupled receptor, 1 | −4.20 | −21.82 | 72 h |
| ENSG00000186074 | NKIR | NK inhibitory receptor precursor | −4.10 | −17.18 | 72 h |
| ENSG00000153266 | ZNF312 | Zinc finger protein 312 | −3.91 | −116.99 | 6 h |
| ENSG00000143595 | AQP10 | Aquaporin 10 | −3.32 | −97.38 | 72 h |
| ENSG00000153266 | ZNF312 | Zinc finger protein 312 | −3.01 | −71.64 | 72 h |
| ENSG00000167183 | PRR15L | Proline rich 15-like | −2.95 | −39.58 | 72 h |
| ENSG00000131470 | TBPIP | TBP-1 interacting protein (TBPIP) | −2.80 | −17.88 | 6 h |
| ENSG00000134389 | CFHL5 | Complement factor H-related 5 | −2.72 | 0.37 | 48 h |
| ENSG00000156475 | PPP2R2B | Protein phosphatase 2, regulatory subunit B (PR 52) | −2.65 | −26.85 | 72 h |
| ENSG00000159261 | CLDN14 | Claudin 14 (CLDN14) | −2.58 | −26.90 | 72 h |
| ENSG00000186910 | SERPINA11 | Serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin) | −2.53 | −25.05 | 72 h |
| ENSG00000182782 | GPR109A | G protein-coupled receptor 109A | −2.50 | −29.16 | 72 h |
| ENSG00000182782 | GPR109B | G protein-coupled receptor 109B | −2.50 | −29.16 | 72 h |
| ENSG00000197711 | HP | Haptoglobin | −2.42 | −291.19 | 72 h |
| ENSG00000197711 | HPR | Haptoglobin-related protein | −2.42 | −291.19 | 72 h |
| ENSG00000130427 | EPO | Erythropoietin | −2.39 | −44.50 | 72 h |
| ENSG00000143224 | PPOX | Protoporphyrinogen oxidase | −2.37 | −15.81 | 6 h |
| ENSG00000162344 | FGF19 | Fibroblast growth factor 19 | −2.31 | −40.79 | 6 h |
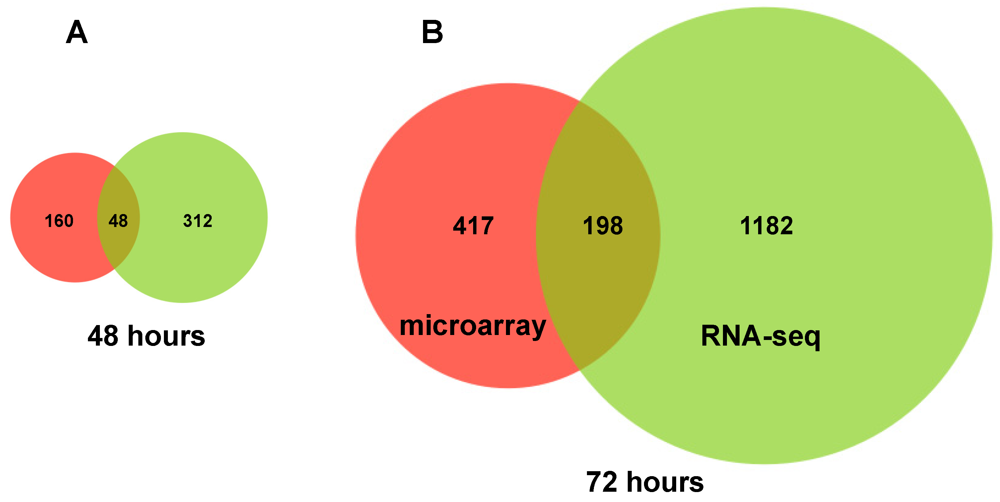
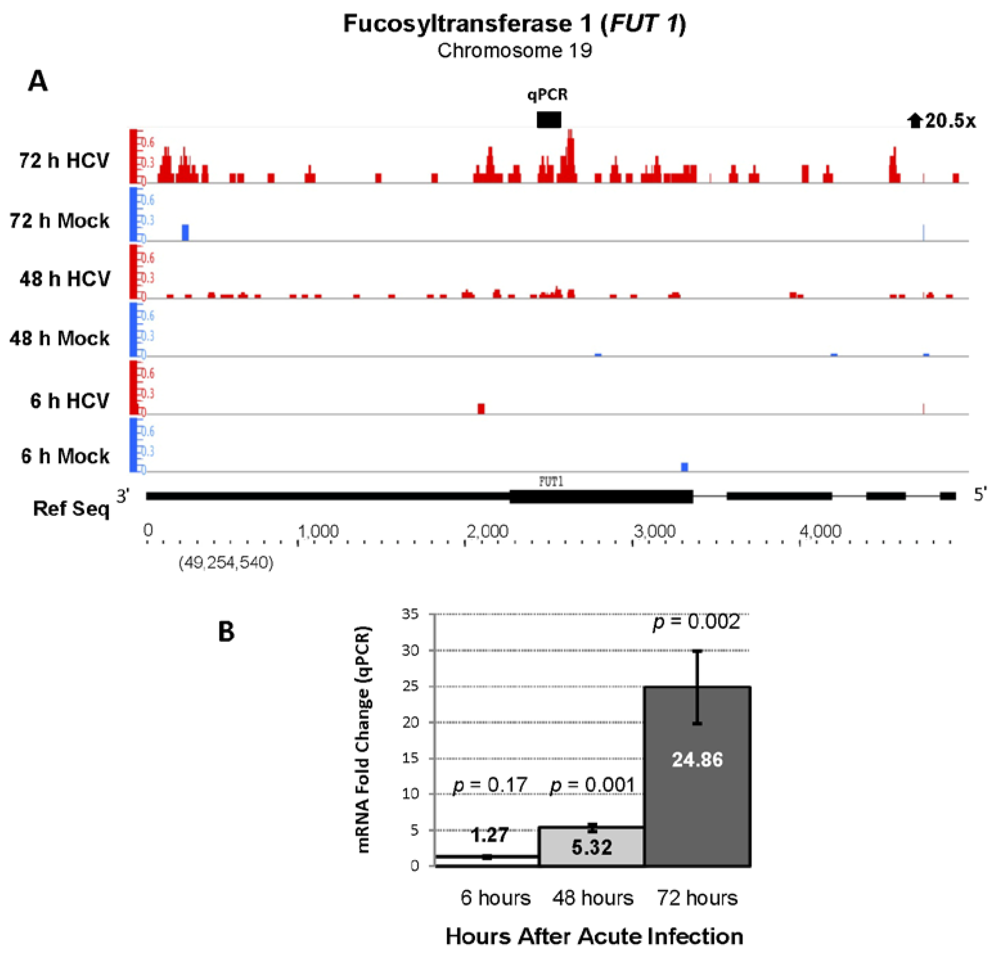
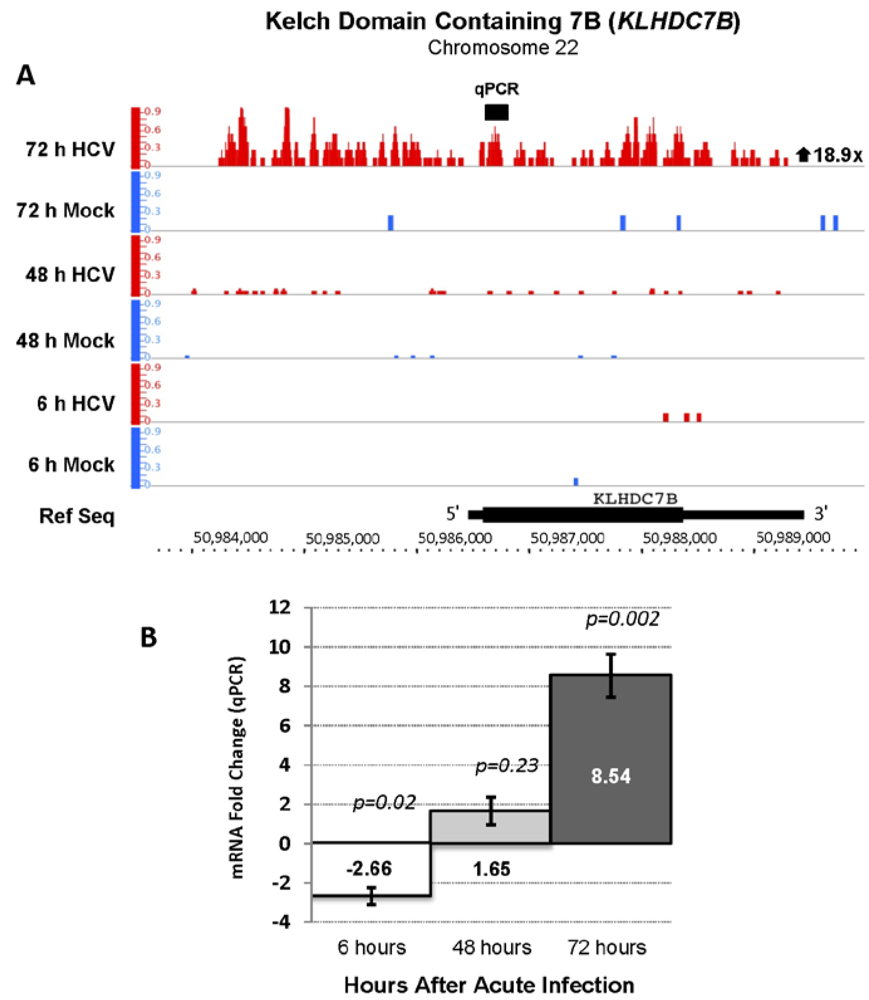
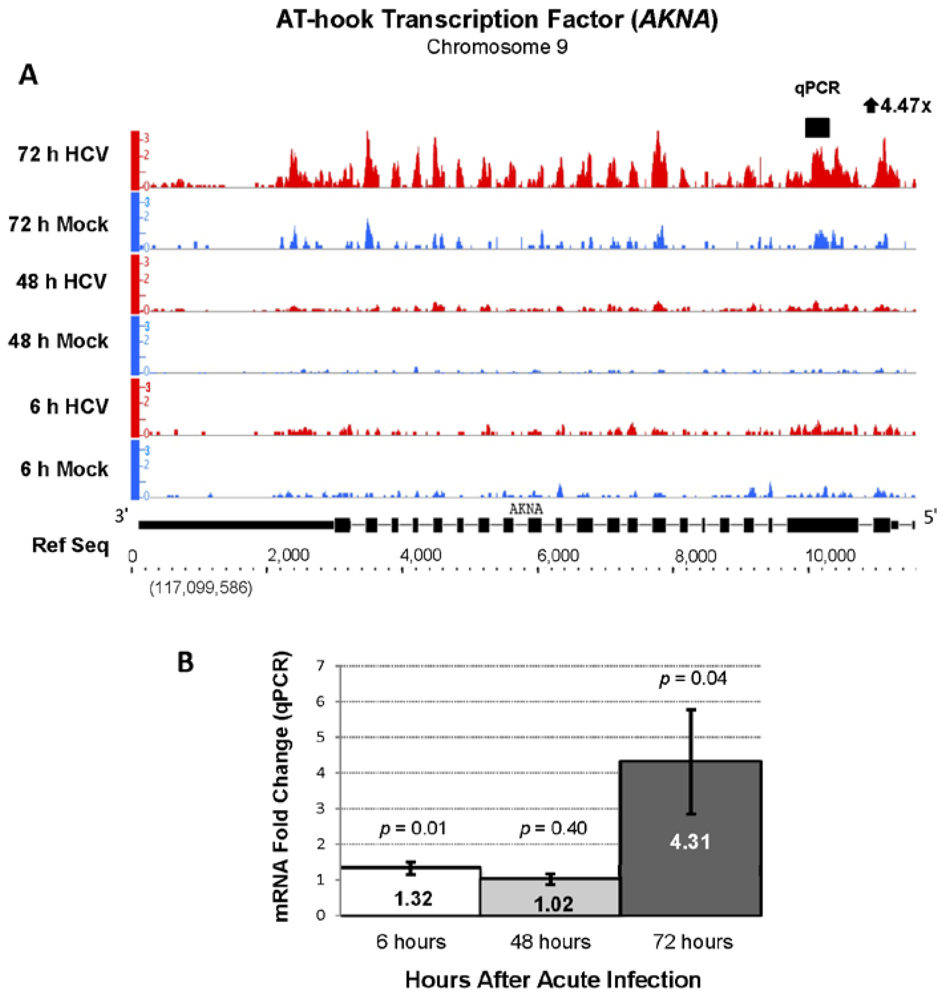
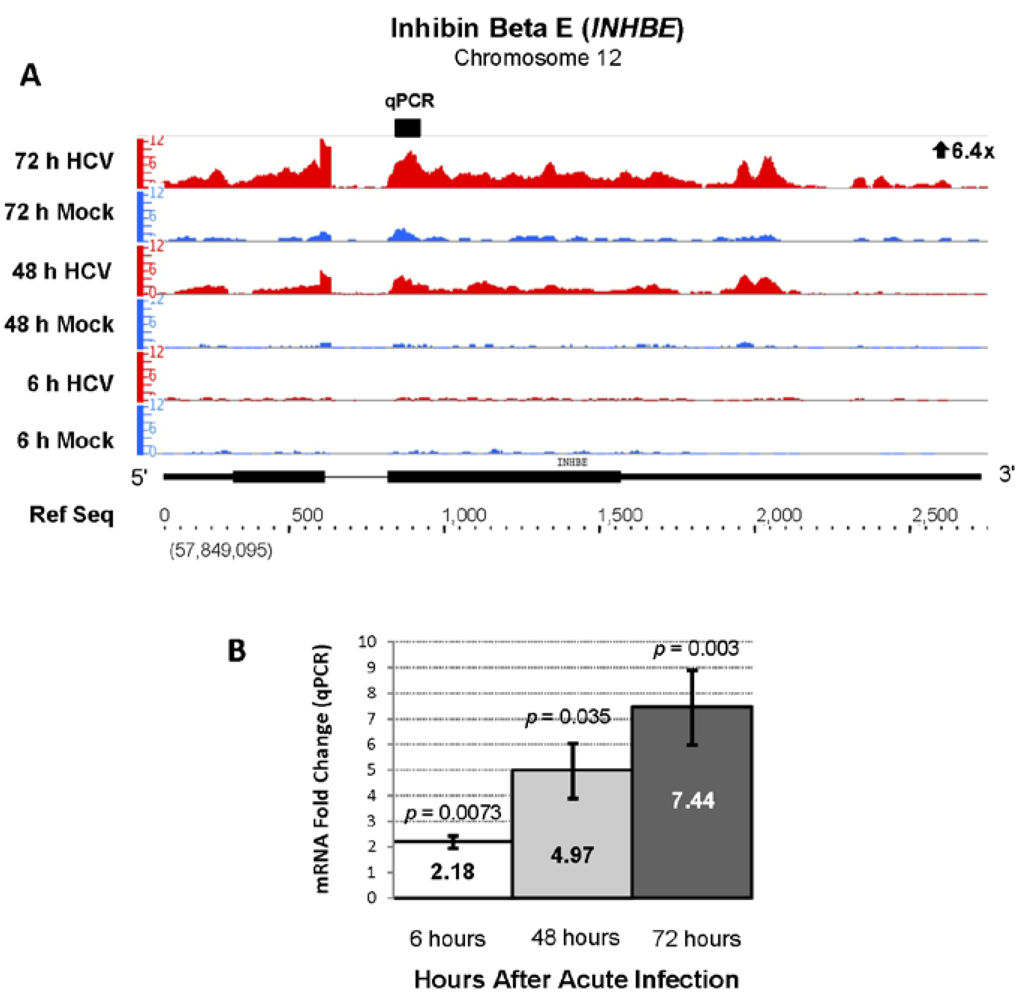
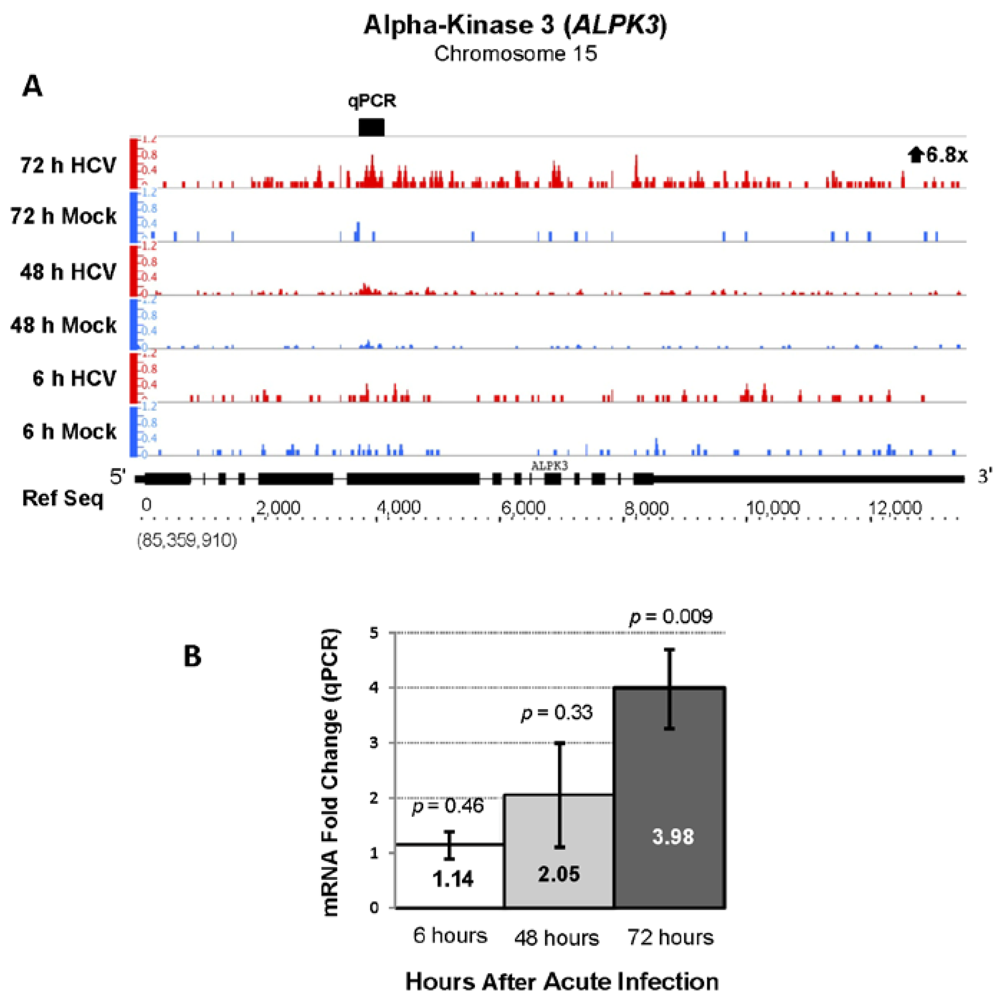
2.3. Changes in Kegg Pathways during Acute HCV Infection
| KEGG Pathway name | Genes in Pathway | 6 h | 48 h | 72 h | |||
|---|---|---|---|---|---|---|---|
| DEGs (EGs) | p value | DEGs (EGs) | p value | DEGs (EGs) | p value | ||
| MAPK signaling | 268 | 5 (168) | 0.003 | 13 (173) | 0.01 | 33 (177) | 0.0005 |
| Cytokine-cytokine receptor interaction | 265 | 9 (90) | 0.005 | 19 (92) | 0.002 | ||
| Antigen processing and presentation | 78 | 2 (36) | 0.02 | ||||
| RIG-I-like receptor signaling | 71 | 12 (44) | 0.001 | ||||
| Toll-like receptor signaling | 102 | 11 (58) | 0.03 | ||||
| NOD-like receptor signaling | 59 | 9 (40) | 0.02 | ||||
| Cytosolic DNA-sensing pathway | 56 | 7 (28) | 0.02 | ||||
| Epithelial cell signaling in H. pylori infection | 68 | 11 (53) | 0.01 | ||||
| Adipocytokine signaling | 68 | 7 (50) | 0.002 | 13 (48) | 0.0008 | ||
| TGF-beta signaling | 85 | 6 (68) | 0.03 | 13 (55) | 0.02 | ||
| Regulation of autophagy | 34 | 5 (18) | 0.03 | ||||
| Apoptosis | 89 | 12 (68) | 0.04 | ||||
| ECM-receptor interaction | 85 | 4 (37) | 0.04 | ||||
| Jak-STAT signaling | 155 | 15 (77) | 0.01 | ||||
| Hepatitis C | 135 | 20 (98) | 0.002 | ||||
| mTOR signaling | 52 | 2 (41) | 0.02 | ||||
| Hedgehog signaling | 56 | 5 (30) | 0.004 | ||||
| Notch signaling | 47 | 9 (37) | 0.01 | ||||
| Renal cell carcinoma | 70 | 2 (58) | 0.04 | ||||
| Basal cell carcinoma | 55 | 4 (31) | 0.02 | ||||
| Hematopoietic cell lineage | 88 | 4 (29) | 0.02 | ||||
| Osteoclast differentiation | 128 | 7 (76) | 0.02 | 19 (76) | 0.0001 | ||
| Insulin signaling | 138 | 19 (110) | 0.01 | ||||
| Type II diabetes mellitus | 48 | 4 (27) | 0.01 | ||||
| Aminoacyl tRNA biosynthesis | 63 | 13 (41) | 0.0001 | ||||
| Ribosome biogenesis in eukaryotes | 81 | 12 (44) | 0.001 | ||||
| Sphingolipid metabolism | 40 | 9 (33) | 0.005 | ||||
| Glutathione metabolism | 50 | 2 (39) | 0.03 | ||||
| Cyanoamino acid metabolism | 7 | 1 (4) | 0.02 | ||||
| Ubiquinone and other terpenoid-quinone biosynthesis | 7 | 1 (5) | 0.03 | ||||
| Taurine and hypotaurine metabolism | 10 | 1 (5) | 0.03 | ||||
| Neurotophin signaling | 127 | 18 (94) | 0.006 | ||||
| One carbon pool by folate | 18 | 5 (17) | 0.03 | ||||
| Prion diseases | 36 | 2 (21) | 0.006 | ||||
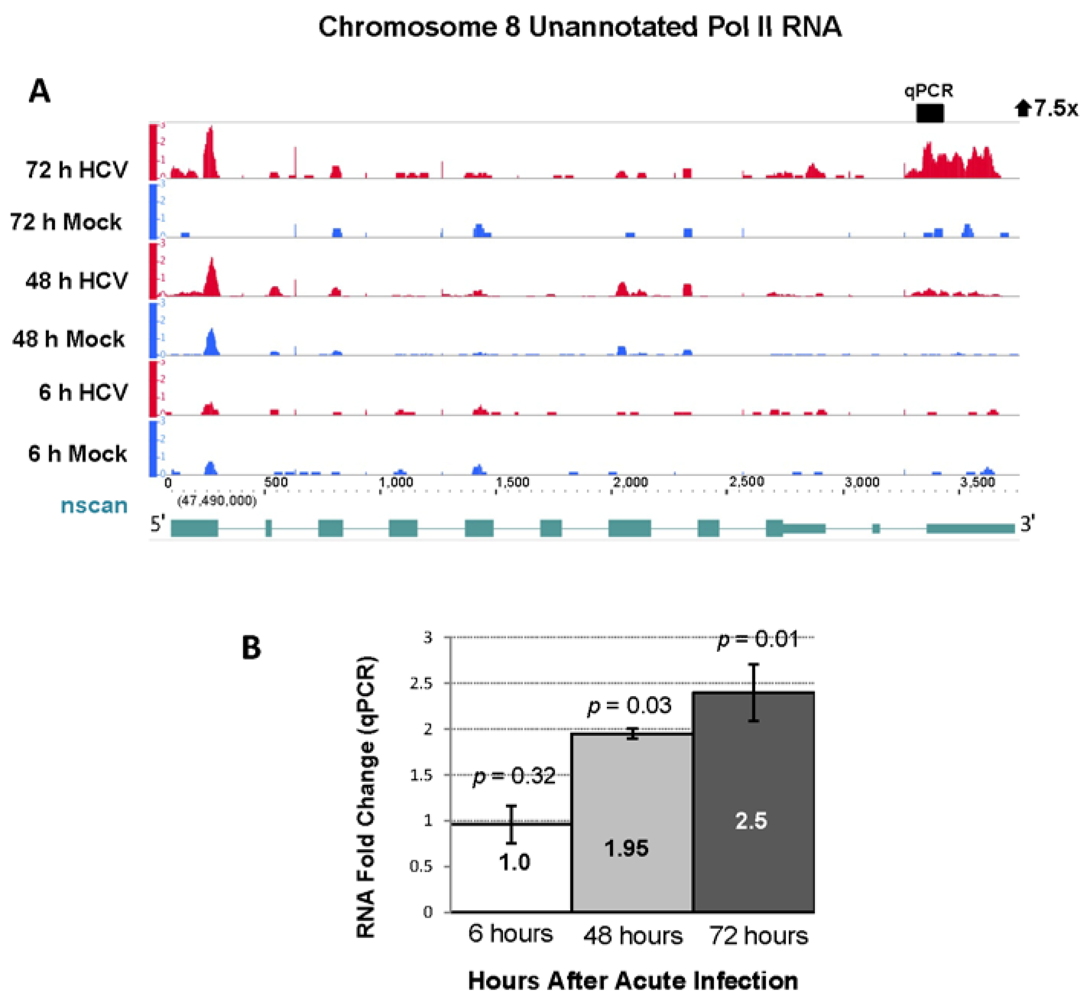
2.4. RNA Sequencing Analysis of 5' Capped RNA Identifies Upregulated Unannotated Pol II Transcripts
| GENOME POSITION | ||||||||
|---|---|---|---|---|---|---|---|---|
| Chr | Start | Stop | Total BPs | HCV RPKM | MOCK RPKM | Fold Change | FDR | Time point |
| 8 | 47528429 | 47529270 | 841 | 7.74 | 0.93 | 7.52 | 113.0 | 72 h |
| 5 | 85865845 | 85866626 | 781 | 10.08 | 3.37 | 2.92 | 57.2 | 72 h |
| 4 | 1384494 | 1385183 | 689 | 6.10 | 2.27 | 2.59 | 21.4 | 72 h |
| 8 | 87354779 | 87355196 | 417 | 7.95 | 3.04 | 2.50 | 14.7 | 72 h |
| 16 | 31580193 | 31580636 | 443 | 10.55 | 4.19 | 2.44 | 21.4 | 72 h |
| 17 | 7967452 | 7968557 | 1105 | 6.15 | 2.62 | 2.33 | 101.1 | 48 h |
| 16 | 21513065 | 21513758 | 693 | 8.71 | 3.94 | 2.17 | 21.9 | 72 h |
| 15 | 64995200 | 64995463 | 263 | 31.29 | 15.07 | 2.07 | 95.1 | 48 h |
| 11 | 122489643 | 122489913 | 270 | 9.48 | 4.76 | 1.97 | 18.1 | 48 h |
| 14 | 65747331 | 65747885 | 554 | 11.64 | 6.17 | 1.86 | 15.4 | 72 h |
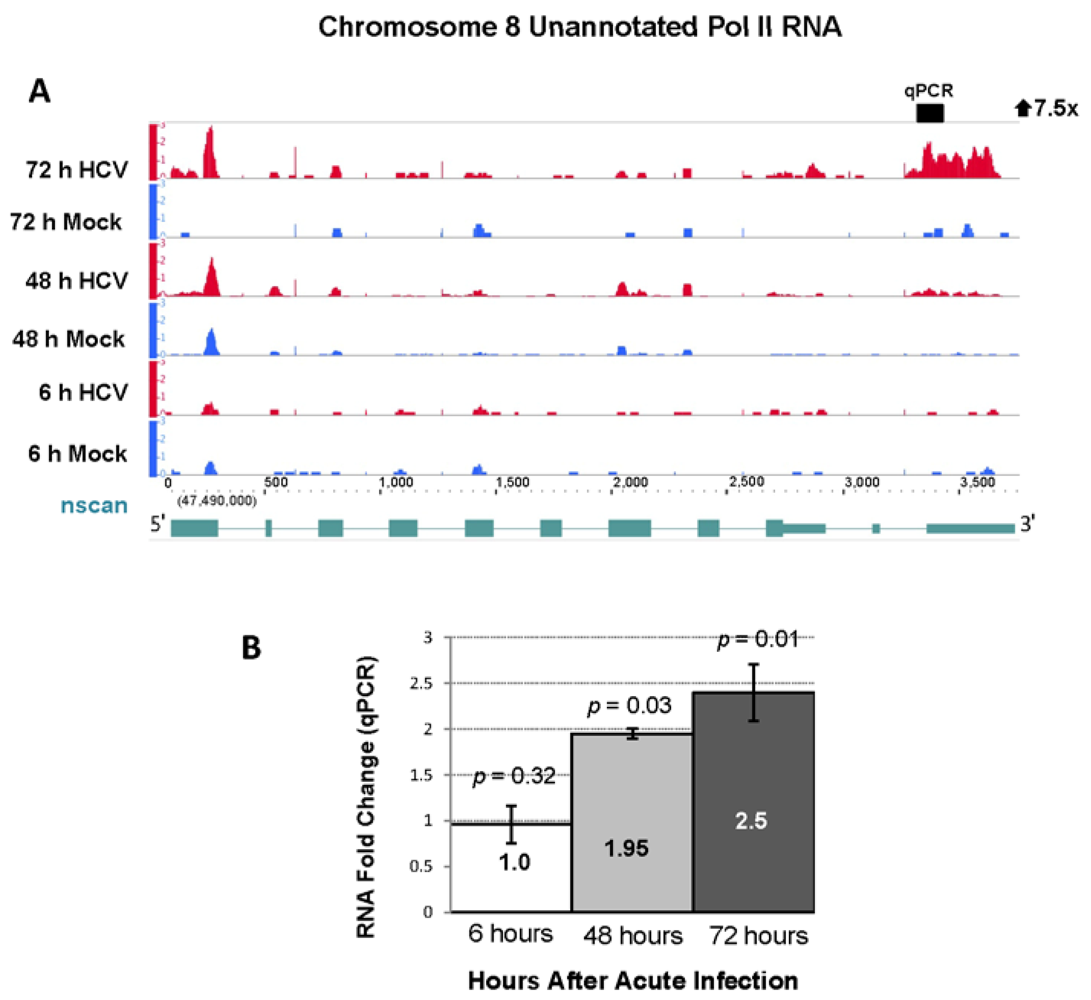
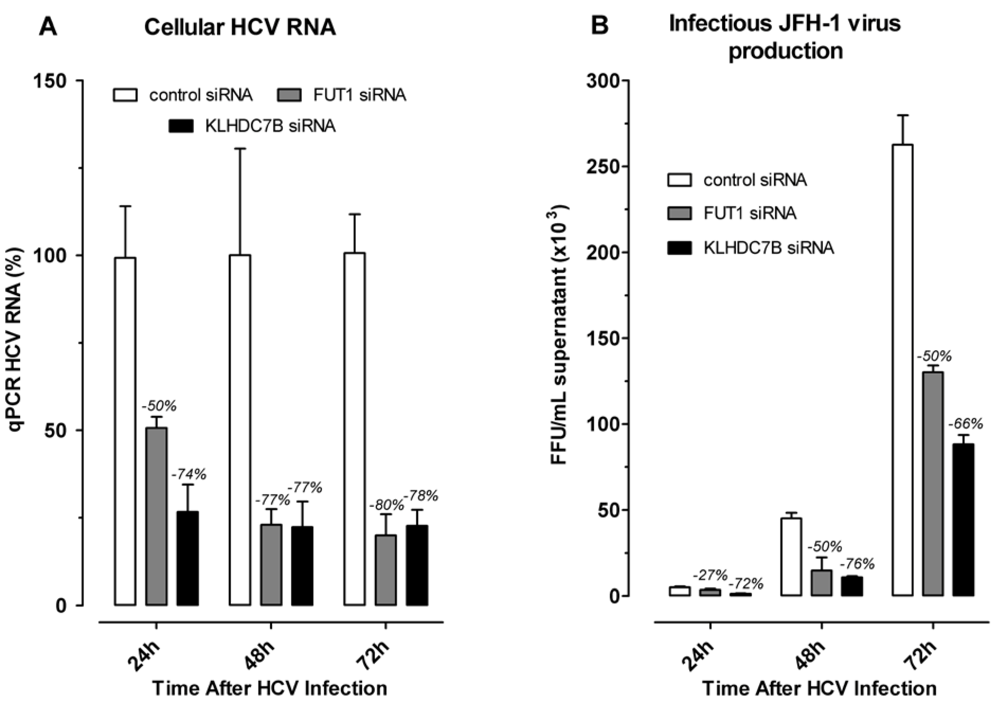
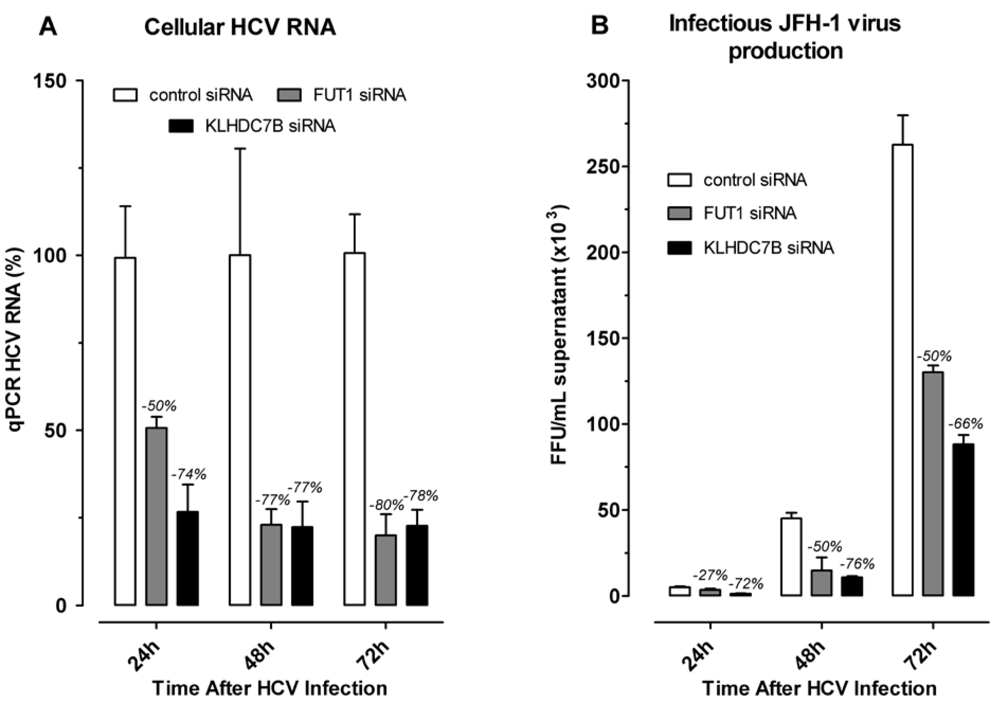
2.4. Effect of Gene Silencing of Selected Upregulated Genes on HCV JFH-1 Replication and Infectious Virus Production
3. Discussion
3.1. Acute HCV Infection Reprograms Gene Transcription of Infected Cells
3.2. Acute HCV Infection Related Kegg Pathways
3.3. HCV and Cellular Glycosylation Pathways
3.4. KLDC7B Gene Silencing Inhibits HCV Replication
4. Experimental Procedures
4.1. Cell Culture
4.2. Production of Infectious HCV
4.3. Infection of Cells with HCV JFH1
4.4. RNA Isolation
4.5. RNA Sequencing
4.6. Bioinformatics
4.7. Real-Time PCR (qPCR)
4.9. RNA Interference
4.10. Quantification of Infectious HCV Virions and HCV RNA
5. Conclusions
Acknowledgments
Funding
Conflict of Interest
References and Notes
- Alter, H.J.; Seeff, L.B. Recovery, persistence, and sequelae in hepatitis C virus infection: A perspective on long-term outcome. Semin. Liver Dis. 2000, 20, 17–35. [Google Scholar] [CrossRef]
- Bialek, S.R.; Terrault, N.A. The changing epidemiology and natural history of hepatitis C virus infection. Clin. Liver Dis. 2006, 10, 697–715. [Google Scholar]
- Murray, C.L.; Rice, C.M. Turning hepatitis C into a real virus. Annu. Rev. Microbiol. 2011, 65, 307–327. [Google Scholar]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for previously treated chronic HCV genotype 1 infection. New Engl. J. Med. 2011, 364, 1207–1217. [Google Scholar]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Goncales, F.L., Jr.; Haussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. New Engl. J. Med. 2002, 347, 975–982. [Google Scholar]
- Poordad, F.; McCone, J., Jr.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for untreated chronic HCV genotype 1 infection. New Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar]
- Shiina, M.; Rehermann, B. Hepatitis C vaccines: Inducing and challenging memory T cells. Hepatology 2006, 43, 1395–1398. [Google Scholar]
- Blackham, S.; Baillie, A.; Al-Hababi, F.; Remlinger, K.; You, S.; Hamatake, R.; McGarvey, M.J. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 2010, 84, 5404–5414. [Google Scholar] [CrossRef]
- Walters, K.A.; Syder, A.J.; Lederer, S.L.; Diamond, D.L.; Paeper, B.; Rice, C.M.; Katze, M.G. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5, e1000269. [Google Scholar]
- Woodhouse, S.D.; Narayan, R.; Latham, S.; Lee, S.; Antrobus, R.; Gangadharan, B.; Luo, S.; Schroth, G.P.; Klenerman, P.; Zitzmann, N. Transcriptome sequencing, microarray, and proteomic analyses reveal cellular and metabolic impact of hepatitis C virus infection in vitro. Hepatology 2010, 52, 443–453. [Google Scholar] [CrossRef]
- Yang, L.; Duff, M.O.; Graveley, B.R.; Carmichael, G.G.; Chen, L.L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12, R16. [Google Scholar]
- Choi, Y.H.; Hagedorn, C.H. Purifying mRNAs with a high-affinity eIF4E mutant identifies the short 3' poly(A) end phenotype. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7033–7038. [Google Scholar]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar]
- Folkers, M.E.; Delker, D.A.; Maxwell, C.I.; Nelson, C.A.; Schwartz, J.J.; Nix, D.A.; Hagedorn, C.H. ENCODE tiling array analysis identifies differentially expressed annotated and novel 5' capped RNAs in hepatitis C infected liver. PLoS One 2011, 6, e14697. [Google Scholar]
- Gowda, M.; Nunes, C.C.; Sailsbery, J.; Xue, M.; Chen, F.; Nelson, C.A.; Brown, D.E.; Oh, Y.; Meng, S.; Mitchell, T.; et al. Genome-wide characterization of methylguanosine-capped and polyadenylated small RNAs in the rice blast fungus Magnaporthe oryzae. Nucleic Acids Res. 2010, 38, 7558–7569. [Google Scholar]
- Oler, A.J.; Alla, R.K.; Roberts, D.N.; Wong, A.; Hollenhorst, P.C.; Chandler, K.J.; Cassiday, P.A.; Nelson, C.A.; Hagedorn, C.H.; Graves, B.J.; et al. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 2010, 17, 620–628. [Google Scholar] [CrossRef]
- Spivak-Kroizman, T.; Friedland, D.E.; De Staercke, C.; Gernert, K.M.; Goss, D.J.; Hagedorn, C.H. Mutations in the S4-H2 loop of eIF4E which increase the affinity for m7GTP. FEBS Lett. 2002, 516, 9–14. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar]
- Peng, X.; Li, Y.; Walters, K.A.; Rosenzweig, E.R.; Lederer, S.L.; Aicher, L.D.; Proll, S.; Katze, M.G. Computational identification of hepatitis C virus associated microRNA-mRNA regulatory modules in human livers. BMC Genom. 2009, 10, 373. [Google Scholar]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 941–946. [Google Scholar]
- Adams, J.; Kelso, R.; Cooley, L. The kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 2000, 10, 17–24. [Google Scholar]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16410–16415. [Google Scholar]
- Ma, W.; Ortiz-Quintero, B.; Rangel, R.; McKeller, M.R.; Herrera-Rodriguez, S.; Castillo, E.F.; Schluns, K.S.; Hall, M.; Zhang, H.; Suh, W.K.; et al. Coordinate activation of inflammatory gene networks, alveolar destruction and neonatal death in AKNA deficient mice. Cell Res. 2011, 21, 1564–1577. [Google Scholar] [CrossRef]
- Moliterno, A.R.; Resar, L.M. AKNA: Another AT-hook transcription factor "hooking-up" with inflammation. Cell Res. 2011, 21, 1528–1530. [Google Scholar]
- Drummond, A.E.; Fuller, P.J. Activin and inhibin, estrogens and NFkappaB, play roles in ovarian tumourigenesis is there crosstalk? Mol. Cell. Endocrinol. 2011. [Google Scholar]
- Bergauer, F.; Bruning, A.; Shabani, N.; Blankenstein, T.; Juckstock, J.; Dian, D.; Mylonas, I. Inhibin/activin-betaE subunit in normal and malignant human cervical tissue and cervical cancer cell lines. J. Mol. Histol. 2009, 40, 353–359. [Google Scholar]
- Bost, A.G.; Venable, D.; Liu, L.; Heinz, B.A. Cytoskeletal requirements for hepatitis C virus (HCV) RNA synthesis in the HCV replicon cell culture system. J. Virol. 2003, 77, 4401–4408. [Google Scholar]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar]
- Ling, L.; Goeddel, D.V. MIP-T3, a novel protein linking tumor necrosis factor receptor-associated factor 3 to the microtubule network. J. Biol. Chem. 2000, 275, 23852–23860. [Google Scholar]
- Lu, H.; Li, W.; Noble, W.S.; Payan, D.; Anderson, D.C. Riboproteomics of the hepatitis C virus internal ribosomal entry site. J. Proteome Res. 2004, 3, 949–957. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar]
- van Baren, M.J.; Koebbe, B.C.; Brent, M.R. Using N-SCAN or TWINSCAN to predict gene structures in genomic DNA sequences. In Current Protocols in Bioinformatics; John Wiley & Sons: Hoboken, NJ, USA, 2007; Chapter 4, Units 4 and 8. [Google Scholar]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 2000, 28, 1102, 1104. [Google Scholar]
- Basak, S.; Pritchard, D.G.; Bhown, A.S.; Compans, R.W. Glycosylation sites of influenza viral glycoproteins: Characterization of tryptic glycopeptides from the A/USSR(H1N1) hemagglutinin glycoprotein. J. Virol. 1981, 37, 549–558. [Google Scholar]
- Stevens, J.; Blixt, O.; Glaser, L.; Taubenberger, J.K.; Palese, P.; Paulson, J.C.; Wilson, I.A. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J. Mol. Biol. 2006, 355, 1143–1155. [Google Scholar]
- Sainz, B., Jr.; Chisari, F.V. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J. Virol. 2006, 80, 10253–10257. [Google Scholar] [CrossRef]
- Geiman, D.E.; Ton-That, H.; Johnson, J.M.; Yang, V.W. Transactivation and growth suppression by the gut-enriched Kruppel-like factor (Kruppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res. 2000, 28, 1106–1113. [Google Scholar]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar]
- Mayer, A.K.; Muehmer, M.; Mages, J.; Gueinzius, K.; Hess, C.; Heeg, K.; Bals, R.; Lang, R.; Dalpke, A.H. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 2007, 178, 3134–3142. [Google Scholar]
- Muller, V.; Viemann, D.; Schmidt, M.; Endres, N.; Ludwig, S.; Leverkus, M.; Roth, J.; Goebeler, M. Candida albicans triggers activation of distinct signaling pathways to establish a proinflammatory gene expression program in primary human endothelial cells. J. Immunol. 2007, 179, 8435–8445. [Google Scholar]
- Tyner, J.W.; Uchida, O.; Kajiwara, N.; Kim, E.Y.; Patel, A.C.; O'Sullivan, M.P.; Walter, M.J.; Schwendener, R.A.; Cook, D.N.; Danoff, T.M.; et al. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 2005, 11, 1180–1187. [Google Scholar]
- Siddiqa, A.; Sims-Mourtada, J.C.; Guzman-Rojas, L.; Rangel, R.; Guret, C.; Madrid-Marina, V.; Sun, Y.; Martinez-Valdez, H. Regulation of CD40 and CD40 ligand by the AT-hook transcription factor AKNA. Nature 2001, 410, 383–387. [Google Scholar]
- Dickson, A.M.; Wilusz, J. Strategies for viral RNA stability: Live long and prosper. Trends Genet. 2011, 27, 286–293. [Google Scholar]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar]
- Murray, K.E.; Roberts, A.W.; Barton, D.J. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 2001, 7, 1126–1141. [Google Scholar]
- Sean, P.; Nguyen, J.H.; Semler, B.L. Altered interactions between stem-loop IV within the 5' noncoding region of coxsackievirus RNA and poly(rC) binding protein 2: Effects on IRES-mediated translation and viral infectivity. Virology 2009, 389, 45–58. [Google Scholar]
- Walter, B.L.; Parsley, T.B.; Ehrenfeld, E.; Semler, B.L. Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J. Virol. 2002, 76, 12008–12022. [Google Scholar]
- Fujimura, K.; Kano, F.; Murata, M. Identification of PCBP2, a facilitator of IRES-mediated translation, as a novel constituent of stress granules and processing bodies. RNA 2008, 14, 425–431. [Google Scholar] [CrossRef]
- Ji, X.; Kong, J.; Liebhaber, S.A. An RNA-protein complex links enhanced nuclear 3' processing with cytoplasmic mRNA stabilization. EMBO J. 2011, 30, 2622–2633. [Google Scholar]
- Parker, R.; Song, H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004, 11, 121–127. [Google Scholar]
- Middelbeek, J.; Clark, K.; Venselaar, H.; Huynen, M.A.; van Leeuwen, F.N. The alpha-kinase family: An exceptional branch on the protein kinase tree. Cell. Mol. Life Sci. 2010, 67, 875–890. [Google Scholar]
- Zilliox, M.J.; Parmigiani, G.; Griffin, D.E. Gene expression patterns in dendritic cells infected with measles virus compared with other pathogens. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3363–3368. [Google Scholar]
- NCBI. Gene. Available online: http://www.ncbi.nlm.nih.gov/gene (accessed on 6 April 2012).
- Ng, M.H.; Ho, T.H.; Kok, K.H.; Siu, K.L.; Li, J.; Jin, D.Y. MIP-T3 Is a Negative Regulator of Innate Type I IFN Response. J. Immunol. 2011, 187, 6473–6482. [Google Scholar]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar]
- Negro, F.; Alaei, M. Hepatitis C virus and type 2 diabetes. World J. Gastroenterol. 2009, 15, 1537–1547. [Google Scholar]
- Kawaguchi, T.; Ide, T.; Taniguchi, E.; Hirano, E.; Itou, M.; Sumie, S.; Nagao, Y.; Yanagimoto, C.; Hanada, S.; Koga, H.; et al. Clearance of HCV improves insulin resistance, beta-cell function, and hepatic expression of insulin receptor substrate 1 and 2. Am. J. Gastroenterol. 2007, 102, 570–576. [Google Scholar]
- Valverde, A.M.; Burks, D.J.; Fabregat, I.; Fisher, T.L.; Carretero, J.; White, M.F.; Benito, M. Molecular mechanisms of insulin resistance in IRS-2-deficient hepatocytes. Diabetes 2003, 52, 2239–2248. [Google Scholar]
- Boissan, M.; Beurel, E.; Wendum, D.; Rey, C.; Lecluse, Y.; Housset, C.; Lacombe, M.L.; Desbois-Mouthon, C. Overexpression of insulin receptor substrate-2 in human and murine hepatocellular carcinoma. Am. J. Pathol. 2005, 167, 869–877. [Google Scholar]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar]
- Pereira Tde, A.; Witek, R.P.; Syn, W.K.; Choi, S.S.; Bradrick, S.; Karaca, G.F.; Agboola, K.M.; Jung, Y.; Omenetti, A.; Moylan, C.A.; et al. Viral factors induce Hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab. Investig. 2010, 90, 1690–1703. [Google Scholar] [CrossRef]
- Philips, G.M.; Chan, I.S.; Swiderska, M.; Schroder, V.T.; Guy, C.; Karaca, G.F.; Moylan, C.; Venkatraman, T.; Feuerlein, S.; Syn, W.K.; et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS One 2011, 6, e23943. [Google Scholar]
- Choi, S.S.; Bradrick, S.; Qiang, G.; Mostafavi, A.; Chaturvedi, G.; Weinman, S.A.; Diehl, A.M.; Jhaveri, R. Up-regulation of Hedgehog pathway is associated with cellular permissiveness for hepatitis C virus replication. Hepatology 2011, 54, 1580–1590. [Google Scholar]
- Nystrom, K.; Grahn, A.; Lindh, M.; Brytting, M.; Mandel, U.; Larson, G.; Olofsson, S. Virus-induced transcriptional activation of host FUT genes associated with neo-expression of Ley in cytomegalovirus-infected and sialyl-Lex in varicella-zoster virus-infected diploid human cells. Glycobiology 2007, 17, 355–366. [Google Scholar]
- Meany, D.L.; Chan, D.W. Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin. Proteonomics 2011, 8, 7. [Google Scholar]
- Miyoshi, E.; Moriwaki, K.; Nakagawa, T. Biological function of fucosylation in cancer biology. J. Biochem. 2008, 143, 725–729. [Google Scholar]
- Becker, D.J.; Lowe, J.B. Fucose: Biosynthesis and biological function in mammals. Glycobiology 2003, 13, 41R–53R. [Google Scholar]
- Nystrom, K.; Norden, R.; Muylaert, I.; Elias, P.; Larson, G.; Olofsson, S. Induction of sialyl-Lex expression by herpes simplex virus type 1 is dependent on viral immediate early RNA-activated transcription of host fucosyltransferase genes. Glycobiology 2009, 19, 847–859. [Google Scholar]
- Cheung, L.S.; Raman, P.S.; Balzer, E.M.; Wirtz, D.; Konstantopoulos, K. Biophysics of selectin-ligand interactions in inflammation and cancer. Phys. Biol. 2011, 8, 015013. [Google Scholar]
- Balcan, E.; Tuglu, I.; Sahin, M.; Toparlak, P. Cell surface glycosylation diversity of embryonic thymic tissues. Acta Histochem. 2008, 110, 14–25. [Google Scholar]
- Earl, L.A.; Bi, S.; Baum, L.G. N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J. Biol. Chem. 2010, 285, 2232–2244. [Google Scholar]
- Moore, G.T.; Brown, S.J.; Winterhalter, A.C.; Lust, M.; Salvaris, E.J.; Selan, C.; Nandurkar, H.H.; Desmond, P.V.; Cowan, P.J.; d'Apice, A.J. Glycosylation changes in hFUT1 transgenic mice increase TCR signaling and apoptosis resulting in thymocyte maturation arrest. Mol. Immunol. 2008, 45, 2401–2410. [Google Scholar]
- NCBI. GEO Profiles. Available online: http://www.ncbi.nlm.nih.gov/geoprofiles (accessed on 6 April 2012).
- Santegoets, L.A.; Seters, M.; Helmerhorst, T.J.; Heijmans-Antonissen, C.; Hanifi-Moghaddam, P.; Ewing, P.C.; van Ijcken, W.F.; van der Spek, P.J.; van der Meijden, W.I.; Blok, L.J. HPV related VIN: Highly proliferative and diminished responsiveness to extracellular signals. Int. J. Canc. 2007, 121, 759–766. [Google Scholar]
- Njau, F.; Geffers, R.; Thalmann, J.; Haller, H.; Wagner, A.D. Restriction of Chlamydia pneumoniae replication in human dendritic cell by activation of indoleamine 2,3-dioxygenase. Microb. Infect. 2009, 11, 1002–1010. [Google Scholar]
- Mezger, M.; Wozniok, I.; Blockhaus, C.; Kurzai, O.; Hebart, H.; Einsele, H.; Loeffler, J. Impact of mycophenolic acid on the functionality of human polymorphonuclear neutrophils and dendritic cells during interaction with Aspergillus fumigatus. Antimicrob. Agents Chemother. 2008, 52, 2644–2646. [Google Scholar]
- Huang, Y.C.; Li, Z.; Hyseni, X.; Schmitt, M.; Devlin, R.B.; Karoly, E.D.; Soukup, J.M. Identification of gene biomarkers for respiratory syncytial virus infection in a bronchial epithelial cell line. Genomic Med. 2008, 2, 113–125. [Google Scholar]
- Sana, T.R.; Janatpour, M.J.; Sathe, M.; McEvoy, L.M.; McClanahan, T.K. Microarray analysis of primary endothelial cells challenged with different inflammatory and immune cytokines. Cytokine 2005, 29, 256–269. [Google Scholar]
- Adams, J.C. Characterization of a Drosophila melanogaster orthologue of muskelin. Gene 2002, 297, 69–78. [Google Scholar]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar]
- Kato, T.; Furusaka, A.; Miyamoto, M.; Date, T.; Yasui, K.; Hiramoto, J.; Nagayama, K.; Tanaka, T.; Wakita, T. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J. Med. Virol. 2001, 64, 334–339. [Google Scholar]
- Liu, S.; Nelson, C.A.; Xiao, L.; Lu, L.; Seth, P.P.; Davis, D.R.; Hagedorn, C.H. Measuring antiviral activity of benzimidazole molecules that alter IRES RNA structure with an infectious hepatitis C virus chimera expressing Renilla luciferase. Antivir. Res. 2011, 89, 54–63. [Google Scholar]
- Friedland, D.E.; Wooten, W.N.; LaVoy, J.E.; Hagedorn, C.H.; Goss, D.J. A mutant of eukaryotic protein synthesis initiation factor eIF4E(K119A) has an increased binding affinity for both m7G cap analogues and eIF4G peptides. Biochemistry 2005, 44, 4546–4550. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar]
- Nicol, J.W.; Helt, G.A.; Blanchard, S.G., Jr.; Raja, A.; Loraine, A.E. The Integrated Genome Browser: Free software for distribution and exploration of genome-scale datasets. Bioinformatics 2009, 25, 2730–2731. [Google Scholar]
- Nix, D.A.; Courdy, S.J.; Boucher, K.M. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 2008, 9, 523. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Papic, N.; Maxwell, C.I.; Delker, D.A.; Liu, S.; Heale, B.S.E.; Hagedorn, C.H. RNA-Sequencing Analysis of 5' Capped RNAs Identifies Many New Differentially Expressed Genes in Acute Hepatitis C Virus Infection. Viruses 2012, 4, 581-612. https://doi.org/10.3390/v4040581
Papic N, Maxwell CI, Delker DA, Liu S, Heale BSE, Hagedorn CH. RNA-Sequencing Analysis of 5' Capped RNAs Identifies Many New Differentially Expressed Genes in Acute Hepatitis C Virus Infection. Viruses. 2012; 4(4):581-612. https://doi.org/10.3390/v4040581
Chicago/Turabian StylePapic, Neven, Christopher I. Maxwell, Don A. Delker, Shuanghu Liu, Bret S. E. Heale, and Curt H. Hagedorn. 2012. "RNA-Sequencing Analysis of 5' Capped RNAs Identifies Many New Differentially Expressed Genes in Acute Hepatitis C Virus Infection" Viruses 4, no. 4: 581-612. https://doi.org/10.3390/v4040581
APA StylePapic, N., Maxwell, C. I., Delker, D. A., Liu, S., Heale, B. S. E., & Hagedorn, C. H. (2012). RNA-Sequencing Analysis of 5' Capped RNAs Identifies Many New Differentially Expressed Genes in Acute Hepatitis C Virus Infection. Viruses, 4(4), 581-612. https://doi.org/10.3390/v4040581