Novel Approaches to Inhibit HIV Entry

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HIV Entry: The Basics
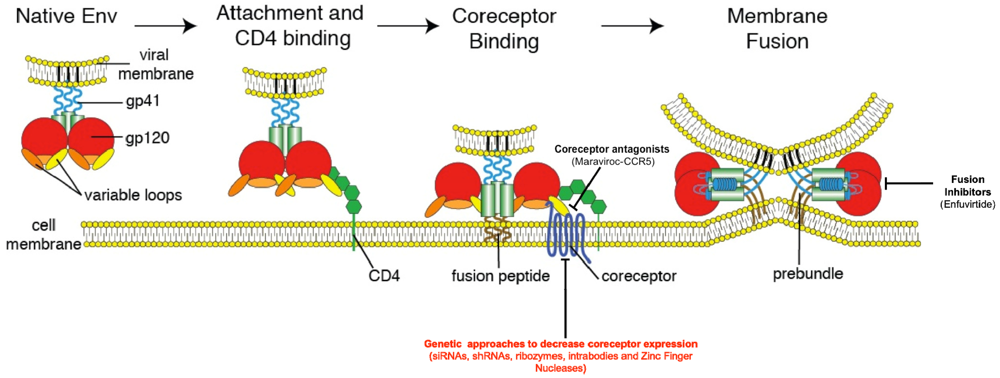
3. Inhibition of HIV Entry
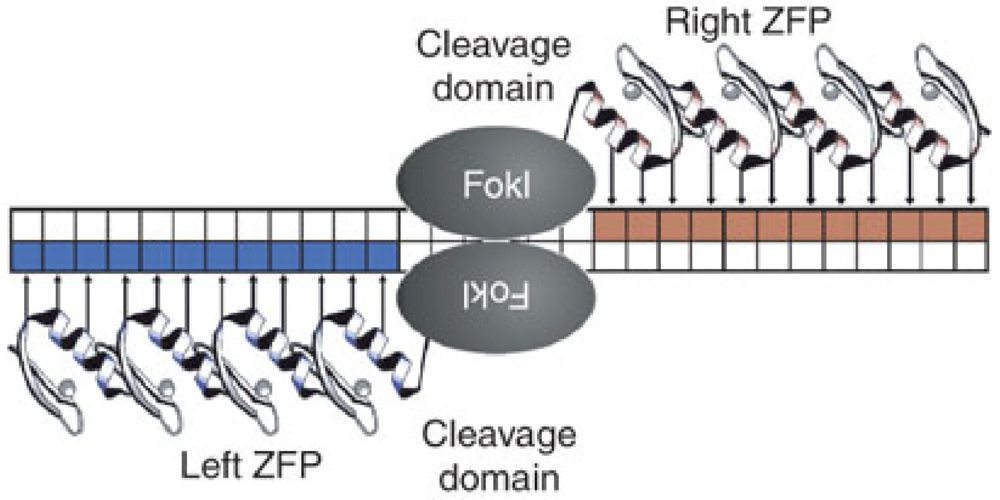
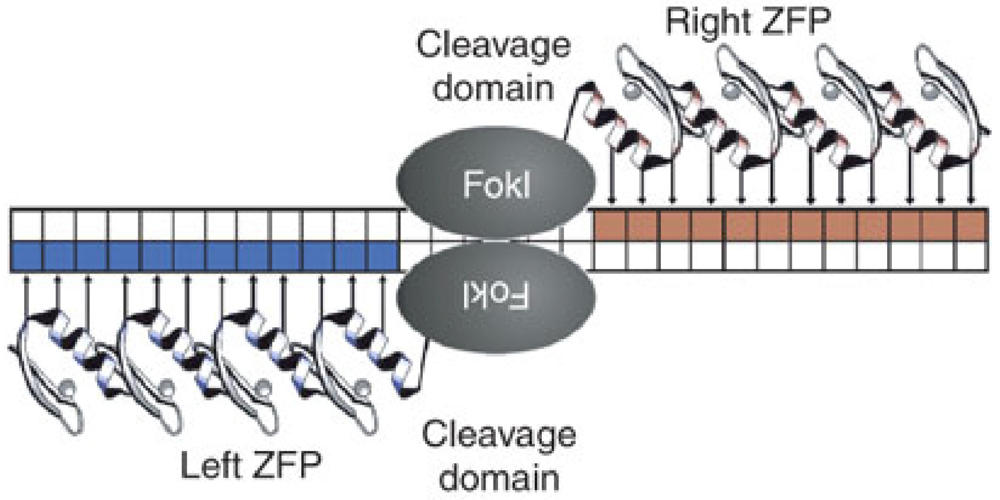
4. Genetic Knockout of CCR5
5. Future Directions
Acknowledgments
Conflict of Interest
References and Notes
- Detels, R.; Munoz, A.; McFarlane, G.; Kingsley, L.A.; Margolick, J.B.; Giorgi, J.; Schrager, L.K.; Phair, J.P. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. JAMA 1998, 280, 1497–1503. [Google Scholar]
- Gulick, R.M.; Meibohm, A.; Havlir, D.; Eron, J.J.; Mosley, A.; Chodakewitz, J.A.; Isaacs, R.; Gonzalez, C.; McMahon, D.; Richman, D.D.; et al. Six-year follow-up of HIV-1-infected adults in a clinical trial of antiretroviral therapy with indinavir, zidovudine, and lamivudine. AIDS 2003, 17, 2345–2349. [Google Scholar]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV Infection, Inflammation, Immunosenescence, and Aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef]
- Maddon, P.J.; Dalgleish, A.G.; McDougal, J.S.; Clapham, P.R.; Weiss, R.A.; Axel, R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 1986, 47, 333–348. [Google Scholar]
- McDougal, J.S.; Maddon, P.J.; Dalgleish, A.G.; Clapham, P.R.; Littman, D.R.; Godfrey, M.; Maddon, D.E.; Chess, L.; Weiss, R.A.; Axel, R. The T4 glycoprotein is a cell-surface receptor for the AIDS virus. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 703–711. [Google Scholar]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996, 85, 1135–1148. [Google Scholar]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar]
- Doranz, B.J.; Rucker, J.; Yi, Y.; Smyth, R.J.; Samson, M.; Peiper, S.C.; Parmentier, M.; Collman, R.G.; Doms, R.W. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 1996, 85, 1149–1158. [Google Scholar] [CrossRef]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996, 382, 833–835. [Google Scholar]
- Schuitemaker, H.; Koot, M.; Kootstra, N.A.; Dercksen, M.W.; de Goede, R.E.; van Steenwijk, R.P.; Lange, J.M.; Schattenkerk, J.K.; Miedema, F.; Tersmette, M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992, 66, 1354–1360. [Google Scholar]
- Roos, M.T.; Lange, J.M.; de Goede, R.E.; Coutinho, R.A.; Schellekens, P.T.; Miedema, F.; Tersmette, M. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J Infect Dis 1992, 165, 427–432. [Google Scholar]
- Zhu, T.; Mo, H.; Wang, N.; Nam, D.S.; Cao, Y.; Koup, R.A.; Ho, D.D. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 1993, 261, 1179–1181. [Google Scholar]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7552–7557. [Google Scholar]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1—Infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar]
- Hunt, P.W.; Harrigan, P.R.; Huang, W.; Bates, M.; Williamson, D.W.; McCune, J.M.; Price, R.W.; Spudich, S.S.; Lampiris, H.; Hoh, R.; et al. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J. Infect. Dis. 2006, 194, 926–930. [Google Scholar]
- Hoffmann, C. The epidemiology of HIV coreceptor tropism. Eur. J. Med. Res. 2007, 12, 385–390. [Google Scholar]
- Koot, M.; Keet, I.P.; Vos, A.H.; de Goede, R.E.; Roos, M.T.; Coutinho, R.A.; Miedema, F.; Schellekens, P.T.; Tersmette, M. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 1993, 118, 681–688. [Google Scholar]
- Huang, Y.; Paxton, W.A.; Wolinsky, S.M.; Neumann, A.U.; Zhang, L.; He, T.; Kang, S.; Ceradini, D.; Jin, Z.; Yazdanbakhsh, K.; et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med. 1996, 2, 1240–1243. [Google Scholar]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5 32/ 32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar]
- H\utter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; M\ussig, A.; Allers, K.; Schneider, T.; Hofmann, J.o.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar]
- Earl, P.L.; Moss, B.; Doms, R.W. Folding, interaction with GRP78-BiP, assembly, and transport of the human immunodeficiency virus type 1 envelope protein. J. Virol. 1991, 65, 2047–2055. [Google Scholar]
- Willey, R.L.; Bonifacino, J.S.; Potts, B.J.; Martin, M.A.; Klausner, R.D. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. PNAS 1988, 85, 9580–9584. [Google Scholar]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.D.; Garten, W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 1992, 360, 358–361. [Google Scholar]
- Reitter, J.N.; Means, R.E.; Desrosiers, R.C. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 1998, 4, 679–684. [Google Scholar]
- Quinones-Kochs, M.I.; Buonocore, L.; Rose, J.K. Role of N-linked glycans in a human immunodeficiency virus envelope glycoprotein: Effects on protein function and the neutralizing antibody response. J. Virol. 2002, 76, 4199–4211. [Google Scholar]
- Fenouillet, E.; Jones, I.; Powell, B.; Schmitt, D.; Kieny, M.P.; Gluckman, J.C. Functional role of the glycan cluster of the human immunodeficiency virus type 1 transmembrane glycoprotein (gp41) ectodomain. J. Virol. 1993, 67, 150–160. [Google Scholar]
- Pohlmann, S.; Baribaud, F.; Doms, R.W. DC-SIGN and DC-SIGNR: Helping hands for HIV. Trends Immun. 2001, 22, 643–646. [Google Scholar]
- Arthos, J.; Cicala, C.; Martinelli, E.; Macleod, K.; Van Ryk, D.; Wei, D.; Xiao, Z.; Veenstra, T.D.; Conrad, T.P.; Lempicki, R.A. HIV-1 envelope protein binds to and signals through integrin α4β7, the gut mucosal homing receptor for peripheral T cells. Nature Immun. 2008, 9, 301–309. [Google Scholar]
- Cicala, C.; Martinelli, E.; McNally, J.P.; Goode, D.J.; Gopaul, R.; Hiatt, J.; Jelicic, K.; Kottilil, S.; Macleod, K.; O'Shea, A.; et al. The integrin alpha4beta7 forms a complex with cell-surface CD4 and defines a T-cell subset that is highly susceptible to infection by HIV-1. Proc. Natl. Acad. Sci. USA 2009, 106, 20877–20882. [Google Scholar]
- Veazey, R.S.; DeMaria, M.; Chalifoux, L.V.; Shvetz, D.E.; Pauley, D.R.; Knight, H.L.; Rosenzweig, M.; Johnson, R.P.; Desrosiers, R.C.; Lackner, A.A. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 1998, 280, 427–431. [Google Scholar]
- Schneider, T.; Ullrich, R.; Zeitz, M. Immunopathology of human immunodeficiency virus infection in the gastrointestinal tract. Springer Semin. Immun. 1997, 18, 515–533. [Google Scholar]
- Clayton, F.; Snow, G.; Reka, S.; Kotler, D.P. Selective depletion of rectal lamina propria rather than lymphoid aggregate CD4 lymphocytes in HIV infection. Clin. Exp. Immun. 1997, 107, 288–292. [Google Scholar]
- Wagner, N.; Lohler, J.; Kunkel, E.J.; Ley, K.; Leung, E.; Krissansen, G.; Rajewsky, K.; Muller, W. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature 1996, 382, 366–370. [Google Scholar]
- Starcich, B.R.; Hahn, B.H.; Shaw, G.M.; McNeely, P.D.; Modrow, S.; Wolf, H.; Parks, E.S.; Parks, W.P.; Josephs, S.F.; Gallo, R.C.; et al. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 1986, 45, 637–648. [Google Scholar]
- Leonard, C.K.; Spellman, M.W.; Riddle, L.; Harris, R.J.; Thomas, J.N.; Gregory, T.J. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 1990, 265, 10373–10382. [Google Scholar]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar]
- DHS NDA 22-128 approval. Available online: http://www.accessdata.fda.gov/drugsatfda_docs /appletter/2007/022128s000ltr.pdf (accessed on 16 January 2012).
- Wu, L.; Gerard, N.P.; Wyatt, R.; Choe, H.; Parolin, C.; Ruffing, N.; Borsetti, A.; Cardoso, A.A.; Desjardin, E.; Newman, W.; et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 1996, 384, 179–183. [Google Scholar]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar]
- Zhang, Y.J.; Dragic, T.; Cao, Y.; Kostrikis, L.; Kwon, D.S.; Littman, D.R.; KewalRamani, V.N.; Moore, J.P. Use of Coreceptors Other Than CCR5 by Non-Syncytium-Inducing Adult and Pediatric Isolates of Human Immunodeficiency Virus Type 1 Is Rare In Vitro. J. Virol. 1998, 72, 9337–9344. [Google Scholar]
- Edinger, A.L.; Clements, J.E.; Doms, R.W. Chemokine and orphan receptors in HIV-2 and SIV tropism and pathogenesis. Virology 1999, 260, 211–221. [Google Scholar]
- Jiang, C.; Parrish, N.F.; Wilen, C.B.; Li, H.; Chen, Y.; Pavlicek, J.W.; Berg, A.; Lu, X.; Song, H.; Tilton, J.C.; et al. Primary infection by a human immunodeficiency virus with atypical coreceptor tropism. J. Virol. 2011, 85, 10669–10681. [Google Scholar]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar]
- van't Wout, A.B.; Kootstra, N.A.; Mulder-Kampinga, G.A.; Albrecht-van Lent, N.; Scherpbier, H.J.; Veenstra, J.; Boer, K.; Coutinho, R.A.; Miedema, F.; Schuitemaker, H. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. The Journal of clinical investigation 1994, 94, 2060–2067. [Google Scholar] [CrossRef]
- Margolis, L.; Shattock, R. Selective transmission of CCR5-utilizing HIV-1: The 'gatekeeper' problem resolved? Nat. Rev. Microbiol. 2006, 4, 312–317. [Google Scholar] [CrossRef]
- Doranz, B.J.; Lu, Z.H.; Rucker, J.; Zhang, T.Y.; Sharron, M.; Cen, Y.H.; Wang, Z.X.; Guo, H.H.; Du, J.G.; Accavitti, M.A.; et al. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J. Virol. 1997, 71, 6305–6314. [Google Scholar]
- Wu, L.; LaRosa, G.; Kassam, N.; Gordon, C.J.; Heath, H.; Ruffing, N.; Chen, H.; Humblias, J.; Samson, M.; Parmentier, M.; et al. Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J. Exp. Med. 1997, 186, 1373–1381. [Google Scholar]
- Trkola, A.; Ketas, T.J.; Nagashima, K.A.; Zhao, L.; Cilliers, T.; Morris, L.; Moore, J.P.; Maddon, P.J.; Olson, W.C. Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J. Viro. 2001, 75, 579–588. [Google Scholar]
- Laakso, M.M.; Lee, F.-H.; Haggarty, B.; Agrawal, C.; Nolan, K.M.; Biscone, M.; Romano, J.; Jordan, A.P.O.; Leslie, G.J.; Meissner, E.G.; et al. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 2007, 3, e117. [Google Scholar]
- Cormier, E.G.; Dragic, T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 2002, 76, 8953–8957. [Google Scholar]
- Huang, C.-C.; Lam, S.N.; Acharya, P.; Tang, M.; Xiang, S.-H.; Hussan, S.S.-U.; Stanfield, R.L.; Robinson, J.; Sodroski, J.; Wilson, I.A.; et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 2007, 317, 1930–1934. [Google Scholar]
- Fouchier, R.A.; Groenink, M.; Kootstra, N.A.; Tersmette, M.; Huisman, H.G.; Miedema, F.; Schuitemaker, H. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 1992, 66, 3183–3187. [Google Scholar]
- De Jong, J.J.; De Ronde, A.; Keulen, W.; Tersmette, M.; Goudsmit, J. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 1992, 66, 6777–6780. [Google Scholar]
- Bosch, M.L.; Earl, P.L.; Fargnoli, K.; Picciafuoco, S.; Giombini, F.; Wongstaal, F.; Franchini, G. Identification of the fusion peptide of primate immunodeficiency viruses. Science 1989, 244, 694–697. [Google Scholar]
- Freed, E.O.; Delwart, E.L.; Buchschacher, G.L.; Panganiban, A.T. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc. Natl. Acad. Sci. USA 1992, 89, 70–74. [Google Scholar]
- Tan, K.; Liu, J.; Wang, J.; Shen, S.; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94, 12303–12308. [Google Scholar]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar]
- Furuta, R.A.; Wild, C.T.; Weng, Y.; Weiss, C.D. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 1998, 5, 276–279. [Google Scholar]
- Markosyan, R.M.; Cohen, F.S.; Melikyan, G.B. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell 2003, 14, 926–938. [Google Scholar]
- McClure, M.O.; Marsh, M.; Weiss, R.A. Human immunodeficiency virus infection of CD4-bearing cells occurs by a pH-independent mechanism. EMBO J. 1988, 7, 513. [Google Scholar]
- Lifson, J.D.; Feinberg, M.B.; Reyes, G.R.; Rabin, L.; Banapour, B.; Chakrabarti, S.; Moss, B.; Wong-Staal, F.; Steimer, K.S.; Engleman, E.G. Induction of CD4-dependent cell fusion by the HTLV-III/LAV envelope glycoprotein. Nature 1986, 323, 725–728. [Google Scholar]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.-G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar]
- Tilton, J.C.; Doms, R.W. Entry inhibitors in the treatment of HIV-1 infection. Antiviral Res. 2010, 85, 91–100. [Google Scholar]
- EMEA EPAR summary for Celsentri (Maraviroc). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_summary_for_the_public/human/000811 /WC500022191.pdf.
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar]
- Lack, N.A.; Green, B.; Dale, D.C.; Calandra, G.B.; Lee, H.; MacFarland, R.T.; Badel, K.; Liles, W.C.; Bridger, G. A pharmacokinetic-pharmacodynamic model for the mobilization of CD34+ hematopoietic progenitor cells by AMD3100. Clin. Pharmacol. Ther. 2005, 77, 427–436. [Google Scholar]
- Liles, W.C.; Rodger, E.; Broxmeyer, H.E.; Dehner, C.; Badel, K.; Calandra, G.; Christensen, J.; Wood, B.; Price, T.H.; Dale, D.C. Augmented mobilization and collection of CD34+ hematopoietic cells from normal human volunteers stimulated with granulocyte-colony-stimulating factor by single-dose administration of AMD3100, a CXCR4 antagonist. Transfusion 2005, 45, 295–300. [Google Scholar]
- Brave, M.; Farrell, A.; Ching Lin, S.; Ocheltree, T.; Pope Miksinski, S.; Lee, S.L.; Saber, H.; Fourie, J.; Tornoe, C.; Booth, B.; et al. FDA review summary: Mozobil in combination with granulocyte colony-stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation. Oncology 2010, 78, 282–288. [Google Scholar]
- Wild, C.; Greenwell, T.; Matthews, T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res. Hum. Retroviruses 1993, 9, 1051–1053. [Google Scholar]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113–113. [Google Scholar]
- Moore, J.P.; Kuritzkes, D.R. A piece de resistance: How HIV-1 escapes small molecule CCR5 inhibitors. Curr. Opin. HIV and AIDS 2009, 4, 118–124. [Google Scholar]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar]
- Melby, T.; Sista, P.; DeMasi, R.; Kirkland, T.; Roberts, N.; Salgo, M.; Heilek-Snyder, G.; Cammack, N.; Matthews, T.J.; Greenberg, M.L. Characterization of Envelope Glycoprotein gp41 Genotype and Phenotypic Susceptibility to Enfuvirtide at Baseline and on Treatment in the Phase III Clinical Trials TORO-1 and TORO-2. AIDS Res. Hum. Retroviruses 2006, 22, 375–385. [Google Scholar]
- H\utter, G.; Zaia, J.A. Allogeneic haematopoietic stem cell transplantation in patients with human immunodeficiency virus: The experiences of more than 25 years. Clin. Exp. Immunol. 2011, 163, 284–295. [Google Scholar]
- Anderson, J.; Li, M.-J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.-K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and Efficacy of a Lentiviral Vector Containing Three Anti-HIV Genes—CCR5 Ribozyme, Tat-rev siRNA, and TAR Decoy—in SCID-hu Mouse–Derived T Cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar]
- Anderson, J.S.; Javien, J.; Nolta, J.A.; Bauer, G. Preintegration HIV-1 Inhibition by a Combination Lentiviral Vector Containing a Chimeric TRIM5α Protein, a CCR5 shRNA, and a TAR Decoy. Mol. Ther. 2009, 17, 2103–2114. [Google Scholar] [CrossRef]
- Liang, M.; Kamata, M.; Chen, K.N. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J. Gene Med. 2010, 12, 1521–2254. [Google Scholar]
- Kim, S.-S.; Peer, D.; Kumar, P.; Subramanya, S.; Wu, H.; Asthana, D.; Habiro, K.; Yang, Y.-G.; Manjunath, N.; Shimaoka, M.; et al. RNAi-mediated CCR5 Silencing by LFA-1-targeted Nanoparticles Prevents HIV Infection in BLT Mice. Mol. Ther. 2009, 18, 370–376. [Google Scholar]
- Swan, C.H.; Buhler, B.; Steinberger, P.; Tschan, M.P.; Barbas, C.F., 3rd; Torbett, B.E. T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 2006, 13, 1480–1492. [Google Scholar]
- Luis Abad, J.; Gonzalez, M.A.; del Real, G.; Mira, E.; Manes, S.; Serrano, F.; Bernad, A. Novel interfering bifunctional molecules against the CCR5 coreceptor are efficient inhibitors of HIV-1 infection. Mol. Ther. 2003, 8, 475–484. [Google Scholar]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-Based Gene Therapy for HIV with Lentiviral Vector-Modified CD34+ Cells in Patients Undergoing Transplantation for AIDS-Related Lymphoma. Sci. Transl. Med. 2010, 2, 36–43. [Google Scholar]
- Mani, M.; Kandavelou, K.; Dy, F.J.; Durai, S.; Chandrasegaran, S. Design, engineering, and characterization of zinc finger nucleases. Biochem. Biophys. Res. Commun. 2005, 335, 447–457. [Google Scholar]
- Podhajska, A.J.; Szybalski, W. Conversion of the FokI endonuclease to a universal restriction enzyme: cleavage of phage M13mp7 DNA at predetermined sites. Gene 1985, 40, 175–182. [Google Scholar]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nature Biotechn. 2007, 25, 778–785. [Google Scholar]
- Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M.H.; Chandrasegaran, S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic. Acids. Res. 2005, 33, 5978–5990. [Google Scholar]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.-L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nature Biotechn. 2008, 26, 808–816. [Google Scholar]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nature Biotechn. 2010, 28, 839–847. [Google Scholar]
- Wilen, C.B.; Wang, J.; Tilton, J.C.; Miller, J.C.; Kim, K.A.; Rebar, E.J.; Sherrill-Mix, S.A.; Patro, S.C.; Secreto, A.J.; Jordan, A.P.O.; et al. Engineering HIV-Resistant Human CD4+ T Cells with CXCR4-Specific Zinc-Finger Nucleases. PLoS Pathog. 2011, 7, e1002020. [Google Scholar]
- Riddick, N.E.; Hermann, E.A.; Loftin, L.M.; Elliott, S.T.; Wey, W.C.; Cervasi, B.; Taaffe, J.; Engram, J.C.; Li, B.; Else, J.G.; et al. A Novel CCR5 Mutation Common in Sooty Mangabeys Reveals SIVsmm Infection of CCR5-Null Natural Hosts and Efficient Alternative Coreceptor Use In Vivo. PLoS Pathog. 2010, 6, e1001064. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Didigu, C.A.; Doms, R.W. Novel Approaches to Inhibit HIV Entry. Viruses 2012, 4, 309-324. https://doi.org/10.3390/v4020309
Didigu CA, Doms RW. Novel Approaches to Inhibit HIV Entry. Viruses. 2012; 4(2):309-324. https://doi.org/10.3390/v4020309
Chicago/Turabian StyleDidigu, Chukwuka A., and Robert W. Doms. 2012. "Novel Approaches to Inhibit HIV Entry" Viruses 4, no. 2: 309-324. https://doi.org/10.3390/v4020309
APA StyleDidigu, C. A., & Doms, R. W. (2012). Novel Approaches to Inhibit HIV Entry. Viruses, 4(2), 309-324. https://doi.org/10.3390/v4020309