The Role of Humoral Innate Immunity in Hepatitis C Virus Infection

Abstract
:1. Virus-Host Interactions in the Acute Phase of HCV Infection
2. The Role of Innate Immunity in Limiting HCV Infection
3. Induction of Acute Phase Proteins in Virus Infections
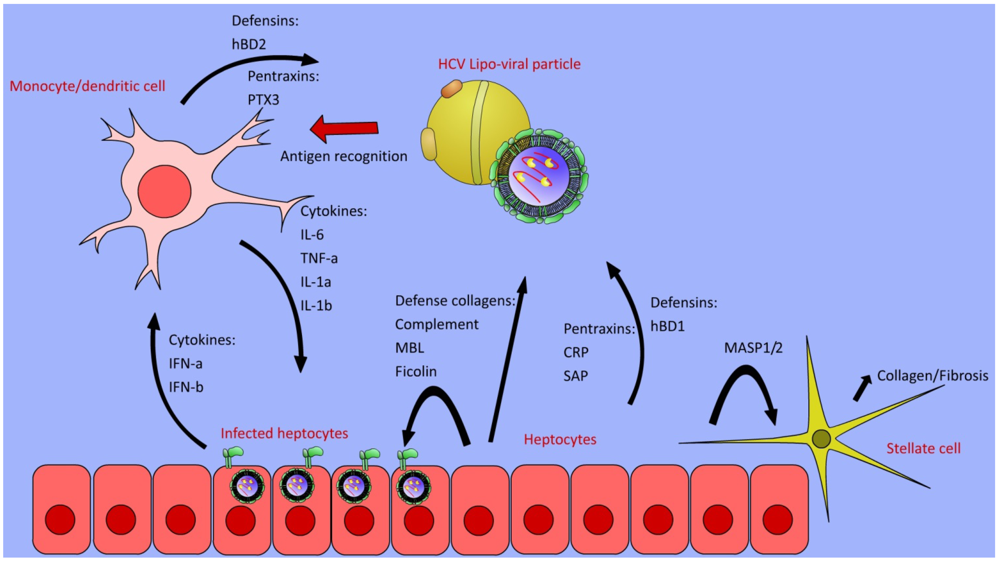
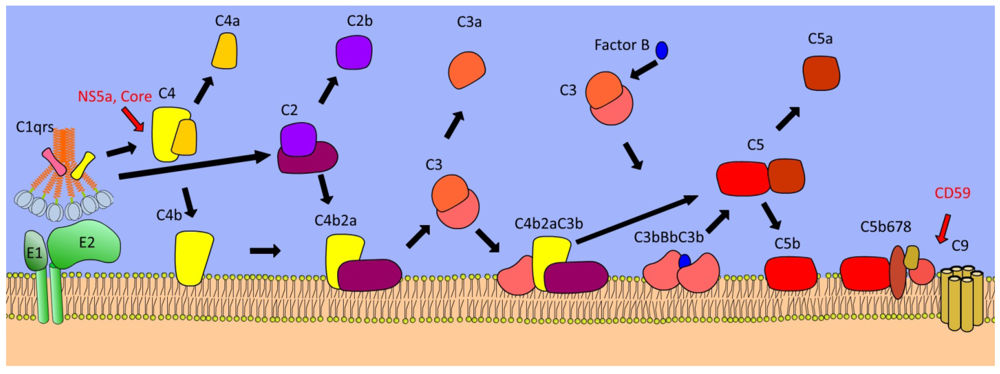
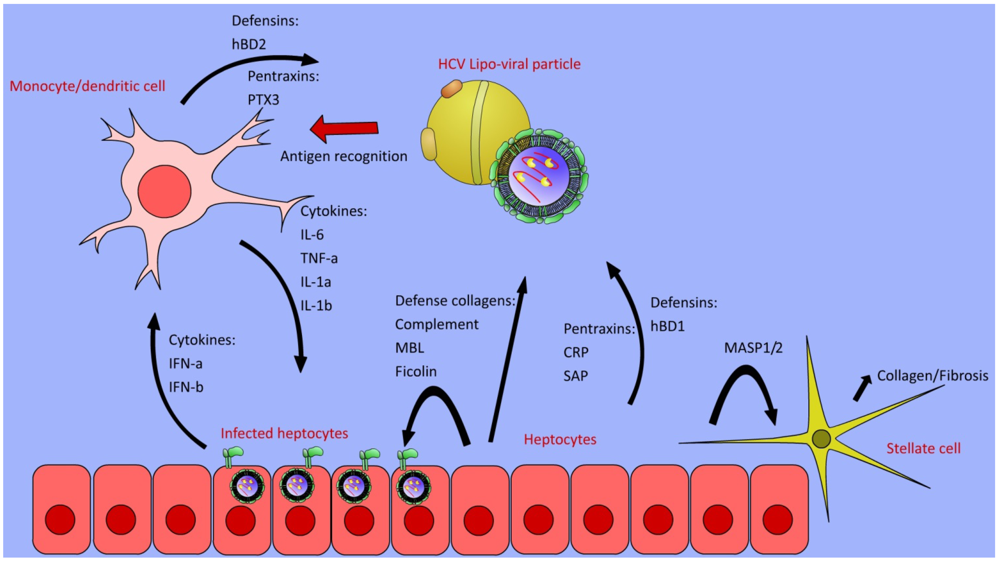
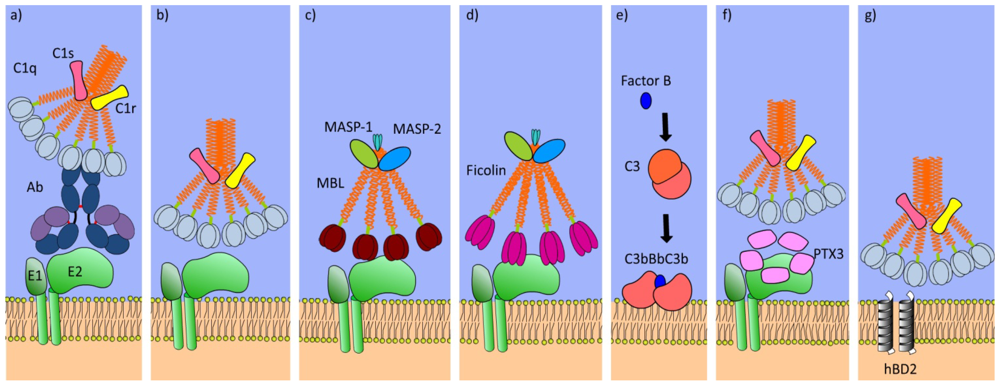
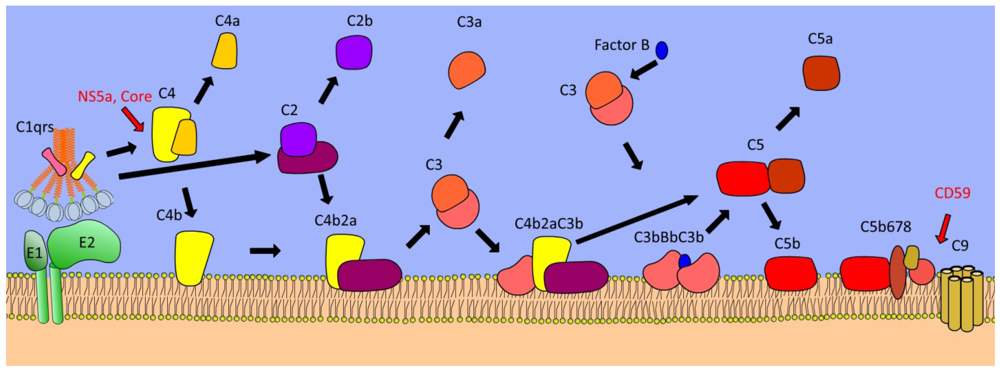
4. Complement Cascade
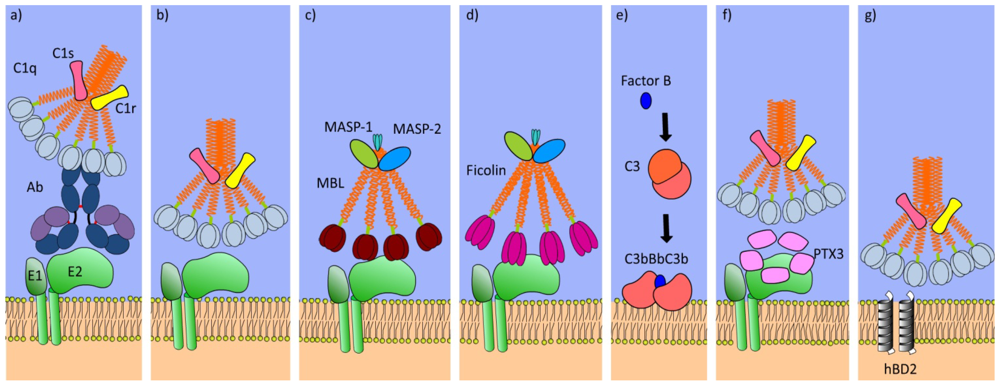
5. Defense Collagens
6. Pentraxins
7. Lipoproteins
8. Defensins
9. Evasion of Immune Responses

{kind=link}
{kind=link}
{kind=link}
| Component | Function | HCV Escape Mechanism | Reference(s) |
|---|---|---|---|
| Complement C4 | Key mediator of opsonisation, direct lysis, and inflammation | Core and NS5a proteins inhibit transcription of C4 | [63] |
| Complement C5-9 | Generation of the membrane attack complex | Incorporation of CD59 into HCV virions | [62] |
| gC1q-R | Enhanced chemotaxis and phagocytosis; decreased activation of B- and T cells | Core interacts with gC1q-R, suppressing T cell activation | [70] |
| Defense collagens (MBL; Ficolins) | Recognition of glycoproteins | HCV glycoproteins have differential glycosylation patterns; association with lipoproteins may block access of defense collagens | [152]; [145] |
| Serum Amyloid A (SAA) | Binding to glycoproteins, inhibiting entry | Interaction with High-density lipoprotein (HDL)/SR-B1 prevents SAA binding? |
10. Role of Innate Immunity in Pathogenesis of HCV Infection
11. Contribution of Innate Immunity to HCV Vaccination and Therapy
12. Concluding Remarks and Future Perspectives
Conflict of interest
Acknowledgements
References and Notes
- Hoofnagle, J.H. Hepatitis C: The clinical spectrum of disease. Hepatology 1997, 26, 15S–20S. [Google Scholar]
- Isaguliants, M.G.; Ozeretskovskaya, N.N. Host background factors contributing to hepatitis C virus clearance. Curr. Pharm. Biotechnol. 2003, 4, 185–193. [Google Scholar]
- Major, M.E.; Dahari, H.; Mihalik, K.; Puig, M.; Rice, C.M.; Neumann, A.U.; Feinstone, S.M. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology 2004, 39, 1709–1720. [Google Scholar]
- Cooper, S.; Erickson, A.L.; Adams, E.J.; Kansopon, J.; Weiner, A.J.; Chien, D.Y.; Houghton, M.; Parham, P.; Walker, C.M. Analysis of a successful immune response against hepatitis C virus. Immunity 1999, 10, 439–449. [Google Scholar]
- Missale, G.; Bertoni, R.; Lamonaca, V.; Valli, A.; Massari, M.; Mori, C.; Rumi, M.G.; Houghton, M.; Fiaccadori, F.; Ferrari, C. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Investig. 1996, 98, 706–714. [Google Scholar]
- Pawlotsky, J.M. Diagnostic tests for hepatitis C. J. Hepatol. 1999, 31, 71–79. [Google Scholar]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15661–15668. [Google Scholar] [PubMed]
- Pape, G.R.; Gerlach, T.J.; Diepolder, H.M.; Gruner, N.; Jung, M.; Santantonio, T. Role of the specific T-cell response for clearance and control of hepatitis C virus. J. Viral. Hepat. 1999, 6, 36–40. [Google Scholar]
- Ishii, K.; Rosa, D.; Watanabe, Y.; Katayama, T.; Harada, H.; Wyatt, C.; Kiyosawa, K.; Aizaki, H.; Matsuura, Y.; Houghton, M.; et al. High titers of antibodies inhibiting the binding of envelope to human cells correlate with natural resolution of chronic hepatitis C. Hepatology 1998, 28, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6025–6030. [Google Scholar] [PubMed]
- Orland, J.R.; Wright, T.L.; Cooper, S. Acute hepatitis C. Hepatology 2001, 33, 321–327. [Google Scholar]
- Spada, E.; Mele, A.; Berton, A.; Ruggeri, L.; Ferrigno, L.; Garbuglia, A.R.; Perrone, M.P.; Girelli, G.; Del Porto, P.; Piccolella, E.; et al. Multispecific T cell response and negative HCV RNA tests during acute HCV infection are early prognostic factors of spontaneous clearance. Gut 2004, 53, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Netski, D.M.; Mosbruger, T.; Depla, E.; Maertens, G.; Ray, S.C.; Hamilton, R.G.; Roundtree, S.; Thomas, D.L.; McKeating, J.; Cox, A. Humoral immune response in acute hepatitis C virus infection. Clin. Infect. Dis. 2005, 41, 667–675. [Google Scholar]
- Dowd, K.A.; Netski, D.M.; Wang, X.H.; Cox, A.L.; Ray, S.C. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 2009, 136, 2377–2386. [Google Scholar]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J.A. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 10149–10154. [Google Scholar]
- Kramer, H.B.; Lavender, K.J.; Qin, L.; Stacey, A.R.; Liu, M.K.; di Gleria, K.; Simmons, A.; Gasper-Smith, N.; Haynes, B.F.; McMichael, A.J.; et al. Elevation of intact and proteolytic fragments of acute phase proteins constitutes the earliest systemic antiviral response in HIV-1 infection. PLoS Pathog. 2010, 6, e1000893. [Google Scholar] [PubMed]
- Barth, H.; Rybczynska, J.; Patient, R.; Choi, Y.; Sapp, R.K.; Baumert, T.F.; Krawczynski, K.; Liang, T.J. Both innate and adaptive immunity mediate protective immunity against hepatitis C virus infection in chimpanzees. Hepatology 2011, 54, 1135–1148. [Google Scholar]
- Crozat, K.; Georgel, P. Identification of mouse cytomegalovirus resistance loci by ENU mutagenesis. Viruses 2009, 1, 460–483. [Google Scholar]
- Crozat, K.; Vivier, E.; Dalod, M. Crosstalk between components of the innate immune system: Promoting anti-microbial defenses and avoiding immunopathologies. Immunol. Rev. 2009, 227, 129–149. [Google Scholar]
- Major, M.E.; Mihalik, K.; Puig, M.; Rehermann, B.; Nascimbeni, M.; Rice, C.M.; Feinstone, S.M. Previously infected and recovered chimpanzees exhibit rapid responses that control hepatitis C virus replication upon rechallenge. J. Virol. 2002, 76, 6586–6595. [Google Scholar]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15669–15674. [Google Scholar] [PubMed]
- Leikina, E.; Delanoe-Ayari, H.; Melikov, K.; Cho, M.S.; Chen, A.; Waring, A.J.; Wang, W.; Xie, Y.; Loo, J.A.; Lehrer, R.I.; et al. Carbohydrate-binding molecules inhibit viral fusion and entry by cross-linking membrane glycoproteins. Nat. Immunol. 2005, 6, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.S.; Keogh, M.J.; Owsianka, A.M.; Adair, R.; Patel, A.H.; Arnold, J.N.; Ball, J.K.; Sim, R.B.; Tarr, A.W.; Hickling, T.P. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell 2010, 1, 664–674. [Google Scholar]
- Buck, C.B.; Day, P.M.; Thompson, C.D.; Lubkowski, J.; Lu, W.; Lowy, D.R.; Schiller, J.T. Human alpha-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1516–1521. [Google Scholar]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar]
- Leibundgut-Landmann, S.; Osorio, F.; Brown, G.D.; Reis e Sousa, C. Stimulation of dendritic cells via the dectin-1/syk pathway allows priming of cytotoxic T-cell responses. Blood 2008, 112, 4971–4980. [Google Scholar]
- Liu, S.; Wu, J.; Zhang, T.; Qian, B.; Wu, P.; Li, L.; Yu, Y.; Cao, X. Complement C1q chemoattracts human dendritic cells and enhances migration of mature dendritic cells to CCL19 via activation of AKT and MAPK pathways. Mol. Immunol. 2008, 46, 242–249. [Google Scholar]
- Joffre, O.; Nolte, M.A.; Sporri, R.; Reis e Sousa, C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol. Rev. 2009, 227, 234–247. [Google Scholar]
- Avirutnan, P.; Mehlhop, E.; Diamond, M.S. Complement and its role in protection and pathogenesis of Flavivirus infections. Vaccine 2008, 26, I100–I107. [Google Scholar]
- Fitzgerald-Bocarsly, P.; Feng, D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007, 89, 843–855. [Google Scholar]
- Newman, K.C.; Riley, E.M. Whatever turns you on: Accessory-cell-dependent activation of NK cells by pathogens. Nat. Rev. Immunol. 2007, 7, 279–291. [Google Scholar]
- Moretta, A.; Marcenaro, E.; Parolini, S.; Ferlazzo, G.; Moretta, L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008, 15, 226–233. [Google Scholar]
- Belz, G.; Mount, A.; Masson, F. Dendritic cells in viral infections. Handb. Exp. Pharmacol. 2009, 51–77. [Google Scholar]
- Perrin-Cocon, L.A.; Villiers, C.L.; Salamero, J.; Gabert, F.; Marche, P.N. B cell receptors and complement receptors target the antigen to distinct intracellular compartments. J. Immunol. 2004, 172, 3564–3572. [Google Scholar]
- Cretin, F.C.; Serra, V.A.; Villiers, M.B.; Laharie, A.M.; Marche, P.N.; Gabert, F.M. C3b complexation diversifies naturally processed T cell epitopes. Mol. Immunol. 2007, 44, 2893–2899. [Google Scholar]
- Barrionuevo, P.; Beigier-Bompadre, M.; Ilarregui, J.M.; Toscano, M.A.; Bianco, G.A.; Isturiz, M.A.; Rabinovich, G.A. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: Galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J. Immunol. 2007, 178, 436–445. [Google Scholar]
- Bayry, J.; Lacroix-Desmazes, S.; Kazatchkine, M.D.; Hermine, O.; Tough, D.F.; Kaveri, S.V. Modulation of dendritic cell maturation and function by B lymphocytes. J. Immunol. 2005, 175, 15–20. [Google Scholar]
- Liu, J.; Ali, M.A.; Shi, Y.; Zhao, Y.; Luo, F.; Yu, J.; Xiang, T.; Tang, J.; Li, D.; Hu, Q.; et al. Specifically binding of L-ficolin to N-glycans of HCV envelope glycoproteins E1 and E2 leads to complement activation. Cell Mol. Immunol. 2009, 6, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Kodys, K.; Kopasz, A.; Marshall, C.; Do, T.; Romics, L., Jr.; Mandrekar, P.; Zapp, M.; Szabo, G. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J. Immunol. 2003, 170, 5615–5624. [Google Scholar]
- Lozach, P.Y.; Lortat-Jacob, H.; de Lacroix de Lavalette, A.; Staropoli, I.; Foung, S.; Amara, A.; Houles, C.; Fieschi, F.; Schwartz, O.; Virelizier, J.L.; et al. DC-sign and L-sign are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 2003, 278, 20358–20366. [Google Scholar] [PubMed]
- Barth, H.; Ulsenheimer, A.; Pape, G.R.; Diepolder, H.M.; Hoffmann, M.; Neumann-Haefelin, C.; Thimme, R.; Henneke, P.; Klein, R.; Paranhos-Baccala, G.; et al. Uptake and presentation of hepatitis C virus-like particles by human dendritic cells. Blood 2005, 105, 3605–3614. [Google Scholar] [PubMed]
- Yamada, E.; Montoya, M.; Schuettler, C.G.; Hickling, T.P.; Tarr, A.W.; Vitelli, A.; Dubuisson, J.; Patel, A.H.; Ball, J.K.; Borrow, P. Analysis of the binding of hepatitis C virus genotype 1a and 1b E2 glycoproteins to peripheral blood mononuclear cell subsets. J. Gen. Virol. 2005, 86, 2507–2512. [Google Scholar]
- Zhou, Y.; Lukes, Y.; Anderson, J.; Fileta, B.; Reinhardt, B.; Sjogren, M. Hepatitis C virus E2 envelope protein induces dendritic cell maturation. J. Viral. Hepat. 2007, 14, 849–858. [Google Scholar]
- Barth, H.; Schnober, E.K.; Neumann-Haefelin, C.; Thumann, C.; Zeisel, M.B.; Diepolder, H.M.; Hu, Z.; Liang, T.J.; Blum, H.E.; Thimme, R.; et al. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J. Virol. 2008, 82, 3466–3479. [Google Scholar] [PubMed]
- Moshage, H. Cytokines and the hepatic acute phase response. J. Pathol. 1997, 181, 257–266. [Google Scholar]
- Gerencer, M.; Burek, V.; Crowe, B.A.; Barrett, N.P.; Dorner, F. The role of complement and gp120-specific antibodies in virus lysis and CD4+ T cell depletion in HIV-1-infected patients. Microb. Pathog. 1998, 25, 253–266. [Google Scholar]
- Sullivan, B.L.; Knopoff, E.J.; Saifuddin, M.; Takefman, D.M.; Saarloos, M.N.; Sha, B.E.; Spear, G.T. Susceptibility of HIV-1 plasma virus to complement-mediated lysis. Evidence for a role in clearance of virus in vivo. J. Immunol. 1996, 157, 1791–1798. [Google Scholar] [PubMed]
- Terajima, M.; Cruz, J.; Co, M.D.; Lee, J.H.; Kaur, K.; Wilson, P.C.; Ennis, F.A. Complement-dependent lysis of influenza A virus-infected cells by broadly cross-reactive human monoclonal antibodies. J. Virol. 2011, 85, 13463–13467. [Google Scholar]
- Hezareh, M.; Hessell, A.J.; Jensen, R.C.; van de Winkel, J.G.; Parren, P.W. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001, 75, 12161–12168. [Google Scholar]
- Gal, P.; Barna, L.; Kocsis, A.; Zavodszky, P. Serine proteases of the classical and lectin pathways: similarities and differences. Immunobiology 2007, 212, 267–277. [Google Scholar]
- Ikeda, F.; Haraguchi, Y.; Jinno, A.; Iino, Y.; Morishita, Y.; Shiraki, H.; Hoshino, H. Human complement component C1q inhibits the infectivity of cell-free HTLV-I. J. Immunol. 1998, 161, 5712–5719. [Google Scholar]
- Ebenbichler, C.F.; Thielens, N.M.; Vornhagen, R.; Marschang, P.; Arlaud, G.J.; Dierich, M.P. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J. Exp. Med. 1991, 174, 1417–1424. [Google Scholar]
- Rus, H.; Cudrici, C.; Niculescu, F. The role of the complement system in innate immunity. Immunol. Res. 2005, 33, 103–112. [Google Scholar]
- Lambris, J.D.; Muller-Eberhard, H.J. The multifunctional role of C3: Structural analysis of its interactions with physiological ligands. Mol. Immunol. 1986, 23, 1237–1242. [Google Scholar]
- Francis, K.; Lewis, B.M.; Akatsu, H.; Monk, P.N.; Cain, S.A.; Scanlon, M.F.; Morgan, B.P.; Ham, J.; Gasque, P. Complement C3a receptors in the pituitary gland: A novel pathway by which an innate immune molecule releases hormones involved in the control of inflammation. FASEB J. 2003, 17, 2266–2268. [Google Scholar]
- Mehlhop, E.; Whitby, K.; Oliphant, T.; Marri, A.; Engle, M.; Diamond, M.S. Complement activation is required for induction of a protective antibody response against West Nile virus infection. J. Virol. 2005, 79, 7466–7477. [Google Scholar]
- Mehlhop, E.; Fuchs, A.; Engle, M.; Diamond, M.S. Complement modulates pathogenesis and antibody-dependent neutralization of West Nile virus infection through a C5-independent mechanism. Virology 2009, 393, 11–15. [Google Scholar]
- Fuchs, A.; Pinto, A.K.; Schwaeble, W.J.; Diamond, M.S. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 2011, 412, 101–109. [Google Scholar]
- Mehlhop, E.; Nelson, S.; Jost, C.A.; Gorlatov, S.; Johnson, S.; Fremont, D.H.; Diamond, M.S.; Pierson, T.C. Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile virus. Cell Host Microbe 2009, 6, 381–391. [Google Scholar]
- Meyer, K.; Basu, A.; Przysiecki, C.T.; Lagging, L.M.; Di Bisceglie, A.M.; Conley, A.J.; Ray, R. Complement-mediated enhancement of antibody function for neutralization of pseudotype virus containing hepatitis C virus E2 chimeric glycoprotein. J. Virol. 2002, 76, 2150–2158. [Google Scholar]
- Dumestre-Perard, C.; Ponard, D.; Drouet, C.; Leroy, V.; Zarski, J.P.; Dutertre, N.; Colomb, M.G. Complement C4 monitoring in the follow-up of chronic hepatitis C treatment. Clin. Exp. Immunol. 2002, 127, 131–136. [Google Scholar]
- Banerjee, A.; Mazumdar, B.; Meyer, K.; Di Bisceglie, A.M.; Ray, R.B.; Ray, R. Transcriptional repression of C4 complement by hepatitis C virus proteins. J. Virol. 2011, 85, 4157–4166. [Google Scholar]
- Amet, T.; Ghabril, M.; Chalasani, N.; Byrd, D.; Hu, N.; Grantham, A.; Liu, Z.; Qin, X.; He, J.J.; Yu, Q. CD59 incorporation protects hepatitis C virus against complement-mediated destruction. Hepatology 2011. [Google Scholar]
- Vanderplasschen, A.; Mathew, E.; Hollinshead, M.; Sim, R.B.; Smith, G.L. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7544–7549. [Google Scholar]
- Spear, G.T.; Lurain, N.S.; Parker, C.J.; Ghassemi, M.; Payne, G.H.; Saifuddin, M. Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV). J. Immunol. 1995, 155, 4376–4381. [Google Scholar] [PubMed]
- Marschang, P.; Sodroski, J.; Wurzner, R.; Dierich, M.P. Decay-accelerating factor (CD55) protects human immunodeficiency virus type 1 from inactivation by human complement. Eur. J. Immunol. 1995, 25, 285–290. [Google Scholar]
- Ghebrehiwet, B.; Lim, B.L.; Peerschke, E.I.; Willis, A.C.; Reid, K.B. Isolation, CDNA cloning, and overexpression of a 33-kd cell surface glycoprotein that binds to the globular "Heads" Of C1q. J. Exp. Med. 1994, 179, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Ghebrehiwet, B.; Lim, B.L.; Kumar, R.; Feng, X.; Peerschke, E.I. gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol. Rev. 2001, 180, 65–77. [Google Scholar] [PubMed]
- Saadoun, D.; Sadallah, S.; Trendelenburg, M.; Limal, N.; Sene, D.; Piette, J.C.; Schifferli, J.A.; Cacoub, P. Anti-C1q antibodies in hepatitis C virus infection. Clin. Exp. Immunol. 2006, 145, 308–312. [Google Scholar]
- Hansen, S.; Holmskov, U. Structural aspects of collectins and receptors for collectins. Immunobiology 1998, 199, 165–189. [Google Scholar]
- Summerfield, J.A.; Ryder, S.; Sumiya, M.; Thursz, M.; Gorchein, A.; Monteil, M.A.; Turner, M.W. Mannose binding protein gene mutations associated with unusual and severe infections in adults. Lancet 1995, 345, 886–889. [Google Scholar]
- Hibberd, M.L.; Sumiya, M.; Summerfield, J.A.; Booy, R.; Levin, M. Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Meningococcal research group. Lancet 1999, 353, 1049–1053. [Google Scholar] [PubMed]
- Ikeda, K.; Sannoh, T.; Kawasaki, N.; Kawasaki, T.; Yamashina, I. Serum lectin with known structure activates complement through the classical pathway. J. Biol. Chem. 1987, 262, 7451–7454. [Google Scholar]
- Ogden, C.A.; deCathelineau, A.; Hoffmann, P.R.; Bratton, D.; Ghebrehiwet, B.; Fadok, V.A.; Henson, P.M. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 2001, 194, 781–795. [Google Scholar]
- Nauta, A.J.; Raaschou-Jensen, N.; Roos, A.; Daha, M.R.; Madsen, H.O.; Borrias-Essers, M.C.; Ryder, L.P.; Koch, C.; Garred, P. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J. Immunol. 2003, 33, 2853–2863. [Google Scholar]
- Thiel, S.; Petersen, S.V.; Vorup-Jensen, T.; Matsushita, M.; Fujita, T.; Stover, C.M.; Schwaeble, W.J.; Jensenius, J.C. Interaction of C1q and mannan-binding lectin (MBL) with C1r, C1s, MBL-associated serine proteases 1 and 2, and the MBL-associated protein Map 19. J. Immunol. 2000, 165, 878–887. [Google Scholar] [PubMed]
- Ambrus, G.; Gal, P.; Kojima, M.; Szilagyi, K.; Balczer, J.; Antal, J.; Graf, L.; Laich, A.; Moffatt, B.E.; Schwaeble, W.; et al. Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: A study on recombinant catalytic fragments. J. Immunol. 2003, 170, 1374–1382. [Google Scholar] [PubMed]
- Gulla, K.C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W.J.; Sim, R.B.; O'Connor, C.D.; Hajela, K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 2009, 129, 482–495. [Google Scholar]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gal, P.; Sim, R.B. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One 2007, 2, e623. [Google Scholar]
- Matsushita, M.; Thiel, S.; Jensenius, J.C.; Terai, I.; Fujita, T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J. Immunol. 2000, 165, 2637–2642. [Google Scholar]
- Hajela, K.; Kojima, M.; Ambrus, G.; Wong, K.H.; Moffatt, B.E.; Ferluga, J.; Hajela, S.; Gal, P.; Sim, R.B. The biological functions of MBL-associated serine proteases (MASPs). Immunobiology 2002, 205, 467–475. [Google Scholar]
- Krarup, A.; Gulla, K.C.; Gal, P.; Hajela, K.; Sim, R.B. The action of MBL-associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta 2008, 1784, 1294–1300. [Google Scholar]
- Megyeri, M.; Mako, V.; Beinrohr, L.; Doleschall, Z.; Prohaszka, Z.; Cervenak, L.; Zavodszky, P.; Gal, P. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J. Immunol. 2009, 183, 3409–3416. [Google Scholar]
- Cortesio, C.L.; Jiang, W. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch. Biochem. Biophys. 2006, 449, 164–170. [Google Scholar]
- Brown, K.S.; Keogh, M.J.; Tagiuri, N.; Grainge, M.J.; Presanis, J.S.; Ryder, S.D.; Irving, W.L.; Ball, J.K.; Sim, R.B.; Hickling, T.P. Severe fibrosis in hepatitis C virus-infected patients is associated with increased activity of the mannan-binding lectin (MBL)/MBL-associated serine protease 1 (MASP-1) complex. Clin. Exp. Immunol. 2007, 147, 90–98. [Google Scholar]
- Terai, I.; Kobayashi, K.; Matsushita, M.; Miyakawa, H.; Mafune, N.; Kikuta, H. Relationship between gene polymorphisms of mannose-binding lectin (MBL) and two molecular forms of MBL. Eur. J. Immunol. 2003, 33, 2755–2763. [Google Scholar]
- Larsen, F.; Madsen, H.O.; Sim, R.B.; Koch, C.; Garred, P. Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J. Biol. Chem. 2004, 279, 21302–21311. [Google Scholar]
- Madsen, H.O.; Garred, P.; Thiel, S.; Kurtzhals, J.A.; Lamm, L.U.; Ryder, L.P.; Svejgaard, A. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J. Immunol. 1995, 155, 3013–3020. [Google Scholar]
- Crosdale, D.J.; Ollier, W.E.; Thomson, W.; Dyer, P.A.; Jensenious, J.; Johnson, R.W.; Poulton, K.V. Mannose binding lectin (MBL) genotype distributions with relation to serum levels in UK Caucasoids. Eur. J. Immunogenet. 2000, 27, 111–117. [Google Scholar]
- Garred, P.; Larsen, F.; Seyfarth, J.; Fujita, R.; Madsen, H.O. Mannose-binding lectin and its genetic variants. Genes Immun. 2006, 7, 85–94. [Google Scholar]
- Matsushita, M.; Hijikata, M.; Ohta, Y.; Iwata, K.; Matsumoto, M.; Nakao, K.; Kanai, K.; Yoshida, N.; Baba, K.; Mishiro, S. Hepatitis C virus infection and mutations of mannose-binding lectin gene MBL. Arch. Virol. 1998, 143, 645–651. [Google Scholar]
- Matsushita, M.; Hijikata, M.; Ohta, Y.; Mishiro, S. Association of mannose-binding lectin gene haplotype LXPA and LYPB with interferon-resistant hepatitis C virus infection in Japanese patients. J. Hepatol. 1998, 29, 695–700. [Google Scholar]
- Sasaki, K.; Tsutsumi, A.; Wakamiya, N.; Ohtani, K.; Suzuki, Y.; Watanabe, Y.; Nakayama, N.; Koike, T. Mannose-binding lectin polymorphisms in patients with hepatitis C virus infection. Scand. J. Gastroenterol. 2000, 35, 960–965. [Google Scholar]
- Koutsounaki, E.; Goulielmos, G.N.; Koulentaki, M.; Choulaki, C.; Kouroumalis, E.; Galanakis, E. Mannose-binding lectin MBL2 gene polymorphisms and outcome of hepatitis C virus-infected patients. J. Clin. Immunol. 2008, 28, 495–500. [Google Scholar]
- Kilpatrick, D.C.; Delahooke, T.E.; Koch, C.; Turner, M.L.; Hayes, P.C. Mannan-binding lectin and hepatitis C infection. Clin. Exp. Immunol. 2003, 132, 92–95. [Google Scholar]
- Iacob, R.E.; Perdivara, I.; Przybylski, M.; Tomer, K.B. Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. J. Am. Soc. Mass. Spectrom. 2008, 19, 428–444. [Google Scholar]
- Worthley, D.L.; Johnson, D.F.; Eisen, D.P.; Dean, M.M.; Heatley, S.L.; Tung, J.P.; Scott, J.; Padbury, R.T.; Harley, H.A.; Bardy, P.G.; et al. Donor mannose-binding lectin deficiency increases the likelihood of clinically significant infection after liver transplantation. Clin. Infect. Dis. 2009, 48, 410–417. [Google Scholar] [PubMed]
- Akaiwa, M.; Yae, Y.; Sugimoto, R.; Suzuki, S.O.; Iwaki, T.; Izuhara, K.; Hamasaki, N. Hakata antigen, a new member of the ficolin/opsonin p35 family, is a novel human lectin secreted into bronchus/alveolus and bile. J. Histochem. Cytochem. 1999, 47, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Keirstead, N.D.; Lee, C.; Yoo, D.; Brooks, A.S.; Hayes, M.A. Porcine plasma ficolin binds and reduces infectivity of porcine reproductive and respiratory syndrome virus (PRRSV) in vitro. Antivir. Res. 2008, 77, 28–38. [Google Scholar]
- Hamed, M.R.; Brown, R.J.P.B.; McClure, C.P.; Urbanowicz, R.A.; Irving, W.L.; Ball, J.K.; Hickling, T.P.; Tarr, A.W. Neutralization of hepatitis C virus entry by human defence collagen L-ficolin. University of Nottingham: Nottingham, UK, 2012; Unpublished work. [Google Scholar]
- Munthe-Fog, L.; Hummelshoj, T.; Hansen, B.E.; Koch, C.; Madsen, H.O.; Skjodt, K.; Garred, P. The impact of FCN2 polymorphisms and haplotypes on the ficolin-2 serum levels. Scand. J. Immunol. 2007, 65, 383–392. [Google Scholar]
- Gulla, K.C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W.J.; Sim, R.B.; O'Connor, C.D.; Hajela, K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 2010, 129, 482–495. [Google Scholar]
- Urban, T.J.; Thompson, A.J.; Bradrick, S.S.; Fellay, J.; Schuppan, D.; Cronin, K.D.; Hong, L.; McKenzie, A.; Patel, K.; Shianna, K.V.; et al. L28b genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology 2010, 52, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Li, J.H.; Thompson, A.; Suchindran, S.; Lao, X.Q.; Patel, K.; Tillmann, H.L.; Muir, A.J.; McHutchison, J.G. Replicated association between an IL28B gene variant and a sustained response to pegylated interferon and ribavirin. Gastroenterology 2010, 138, 2307–2314. [Google Scholar]
- Gewurz, H.; Zhang, X.H.; Lint, T.F. Structure and function of the pentraxins. Curr. Opin. Immunol. 1995, 7, 54–64. [Google Scholar]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Invest. 2003, 111, 1805–1812. [Google Scholar]
- Baruah, P.; Dumitriu, I.E.; Peri, G.; Russo, V.; Mantovani, A.; Manfredi, A.A.; Rovere-Querini, P. The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J. Leukoc. Biol. 2006, 80, 87–95. [Google Scholar]
- Kessel, A.; Elias, G.; Pavlotzky, E.; Zuckerman, E.; Rosner, I.; Toubi, E. Anti-C-reactive protein antibodies in chronic hepatitis C infection: Correlation with severity and autoimmunity. Hum. Immunol. 2007, 68, 844–848. [Google Scholar]
- Alles, V.V.; Bottazzi, B.; Peri, G.; Golay, J.; Introna, M.; Mantovani, A. Inducible expression of PTX3, a new member of the pentraxin family, in human mononuclear phagocytes. Blood 1994, 84, 3483–3493. [Google Scholar] [PubMed]
- Reading, P.C.; Bozza, S.; Gilbertson, B.; Tate, M.; Moretti, S.; Job, E.R.; Crouch, E.C.; Brooks, A.G.; Brown, L.E.; Bottazzi, B.; et al. Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. J. Immunol. 2008, 180, 3391–3398. [Google Scholar] [PubMed]
- Bozza, S.; Bistoni, F.; Gaziano, R.; Pitzurra, L.; Zelante, T.; Bonifazi, P.; Perruccio, K.; Bellocchio, S.; Neri, M.; Iorio, A.M.; et al. Pentraxin 3 protects from MCMV infection and reactivation through TLR sensing pathways leading to IRF3 activation. Blood 2006, 108, 3387–3396. [Google Scholar] [PubMed]
- Nauta, A.J.; Bottazzi, B.; Mantovani, A.; Salvatori, G.; Kishore, U.; Schwaeble, W.J.; Gingras, A.R.; Tzima, S.; Vivanco, F.; Egido, J.; et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur. J. Immunol. 2003, 33, 465–473. [Google Scholar] [PubMed]
- Peri, G.; Introna, M.; Corradi, D.; Iacuitti, G.; Signorini, S.; Avanzini, F.; Pizzetti, F.; Maggioni, A.P.; Moccetti, T.; Metra, M.; et al. PTX3, a prototypical long pentraxin, is an early indicator of acute myocardial infarction in humans. Circulation 2000, 102, 636–641. [Google Scholar] [PubMed]
- Baruah, P.; Propato, A.; Dumitriu, I.E.; Rovere-Querini, P.; Russo, V.; Fontana, R.; Accapezzato, D.; Peri, G.; Mantovani, A.; Barnaba, V.; et al. The pattern recognition receptor PTX3 is recruited at the synapse between dying and dendritic cells, and edits the cross-presentation of self, viral, and tumor antigen. Blood 2006, 107, 151–158. [Google Scholar] [PubMed]
- Jensen, L.E.; Whitehead, A.S. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem. J. 1998, 334, 489–503. [Google Scholar]
- Smith, J.W.; McDonald, T.L. Production of serum amyloid A and C-reactive protein by HepG2 cells stimulated with combinations of cytokines or monocyte conditioned media: The effects of prednisolone. Clin. Exp. Immunol. 1992, 90, 293–299. [Google Scholar]
- Song, C.; Hsu, K.; Yamen, E.; Yan, W.; Fock, J.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem. 2005, 280, 8031–8040. [Google Scholar] [PubMed]
- van der Westhuyzen, D.R.; Cai, L.; de Beer, M.C.; de Beer, F.C. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J. Biol. Chem. 2005, 280, 35890–35895. [Google Scholar]
- Marsche, G.; Frank, S.; Raynes, J.G.; Kozarsky, K.F.; Sattler, W.; Malle, E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem. J. 2007, 402, 117–124. [Google Scholar]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A.; et al. Role of scavenger receptor class B type I in hepatitis C virus entry: Kinetics and molecular determinants. J. Virol. 2009, 84, 34–43. [Google Scholar]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 2005, 79, 8217–8229. [Google Scholar] [PubMed]
- Voisset, C.; Callens, N.; Blanchard, E.; Op De Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar]
- Meunier, J.C.; Engle, R.E.; Faulk, K.; Zhao, M.; Bartosch, B.; Alter, H.; Emerson, S.U.; Cosset, F.L.; Purcell, R.H.; Bukh, J. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4560–4565. [Google Scholar]
- Lavie, M.; Voisset, C.; Vu-Dac, N.; Zurawski, V.; Duverlie, G.; Wychowski, C.; Dubuisson, J. Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology 2006, 44, 1626–1634. [Google Scholar]
- Cai, Z.; Cai, L.; Jiang, J.; Chang, K.S.; van der Westhuyzen, D.R.; Luo, G. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J. Virol. 2007, 81, 6128–6133. [Google Scholar]
- Miwata, H.; Yamada, T.; Okada, M.; Kudo, T.; Kimura, H.; Morishima, T. Serum amyloid A protein in acute viral infections. Arch. Dis. Child. 1993, 68, 210–214. [Google Scholar]
- Norata, G.D.; Pirillo, A.; Ammirati, E.; Catapano, A.L. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis 2012, 220, 11–21. [Google Scholar]
- Klotman, M.E.; Chang, T.L. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar]
- Martin, E.; Ganz, T.; Lehrer, R.I. Defensins and other endogenous peptide antibiotics of vertebrates. J. Leukoc. Biol. 1995, 58, 128–136. [Google Scholar]
- Seidel, A.; Ye, Y.; de Armas, L.R.; Soto, M.; Yarosh, W.; Marcsisin, R.A.; Tran, D.; Selsted, M.E.; Camerini, D. Cyclic and acyclic defensins inhibit human immunodeficiency virus type-1 replication by different mechanisms. PLoS One 2011, 5, e9737. [Google Scholar]
- Prohaszka, Z.; Nemet, K.; Csermely, P.; Hudecz, F.; Mezo, G.; Fust, G. Defensins purified from human granulocytes bind C1q and activate the classical complement pathway like the transmembrane glycoprotein gp41 of HIV-1. Mol. Immunol. 1997, 34, 809–816. [Google Scholar]
- Ryan, L.K.; Dai, J.; Yin, Z.; Megjugorac, N.; Uhlhorn, V.; Yim, S.; Schwartz, K.D.; Abrahams, J.M.; Diamond, G.; Fitzgerald-Bocarsly, P. Modulation of human beta-defensin-1 (hBD-1) in plasmacytoid dendritic cells (pDC), monocytes, and epithelial cells by influenza virus, herpes simplex virus, and sendai virus and its possible role in innate immunity. J. Leukoc. Biol. 2011, 90, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, A.; Skrygan, M.; Gambichler, T.; Brockmeyer, N.H.; Stucker, M.; Herzler, C.; Potthoff, A.; Altmeyer, P.; Pfister, H.; Wieland, U. Human papillomavirus-associated induction of human beta-defensins in anal intraepithelial neoplasia. Br. J. Dermatol. 2009, 160, 1197–1205. [Google Scholar]
- Bhat, S.; Song, Y.H.; Lawyer, C.; Milner, S.M. Modulation of the complement system by human beta-defensin 2. J. Burns Wounds 2007, 5, e10. [Google Scholar]
- Zapata, W.; Rodriguez, B.; Weber, J.; Estrada, H.; Quinones-Mateu, M.E.; Zimermman, P.A.; Lederman, M.M.; Rugeles, M.T. Increased levels of human beta-defensins mRNA in sexually HIV‑1 exposed but uninfected individuals. Curr. HIV Res. 2008, 6, 531–538. [Google Scholar]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar]
- Aceti, A.; Mangoni, M.L.; Pasquazzi, C.; Fiocco, D.; Marangi, M.; Miele, R.; Zechini, B.; Borro, M.; Versace, I.; Simmaco, M. Alpha-defensin increase in peripheral blood mononuclear cells from patients with hepatitis C virus chronic infection. J. Viral. Hepat. 2006, 13, 821–827. [Google Scholar]
- Prince, A.M.; Huima-Byron, T.; Parker, T.S.; Levine, D.M. Visualization of hepatitis C virions and putative defective interfering particles isolated from low-density lipoproteins. J. Viral. Hepat. 1996, 3, 11–17. [Google Scholar]
- Thomssen, R.; Bonk, S.; Thiele, A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 1993, 182, 329–334. [Google Scholar]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar]
- Meunier, J.C.; Russell, R.S.; Engle, R.E.; Faulk, K.N.; Purcell, R.H.; Emerson, S.U. Apolipoprotein C1 association with hepatitis C virus. J. Virol. 2008, 82, 9647–9656. [Google Scholar]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar]
- Tao, W.; Xu, C.; Ding, Q.; Li, R.; Xiang, Y.; Chung, J.; Zhong, J. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology 2009, 395, 67–76. [Google Scholar]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J.A. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar]
- Falkowska, E.; Kajumo, F.; Garcia, E.; Reinus, J.; Dragic, T. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 2007, 81, 8072–8079. [Google Scholar]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.; Keck, Z.Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar]
- Brown, R.J.; Tarr, A.W.; McClure, C.P.; Juttla, V.S.; Tagiuri, N.; Irving, W.L.; Ball, J.K. Cross-genotype characterization of genetic diversity and molecular adaptation in hepatitis C virus envelope glycoprotein genes. J. Gen. Virol. 2007, 88, 458–469. [Google Scholar]
- Farci, P.; Shimoda, A.; Coiana, A.; Diaz, G.; Peddis, G.; Melpolder, J.C.; Strazzera, A.; Chien, D.Y.; Munoz, S.J.; Balestrieri, A.; et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 2000, 288, 339–344. [Google Scholar] [PubMed]
- Job, E.R.; Deng, Y.M.; Tate, M.D.; Bottazzi, B.; Crouch, E.C.; Dean, M.M.; Mantovani, A.; Brooks, A.G.; Reading, P.C. Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J. Immunol. 2010, 185, 4284–4291. [Google Scholar]
- Qin, X.; Gao, B. The complement system in liver diseases. Cell Mol. Immunol. 2006, 3, 333–340. [Google Scholar]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar]
- Levy, M.T.; McCaughan, G.W.; Marinos, G.; Gorrell, M.D. Intrahepatic expression of the hepatic stellate cell marker fibroblast activation protein correlates with the degree of fibrosis in hepatitis C virus infection. Liver 2002, 22, 93–101. [Google Scholar]
- Gaca, M.D.; Zhou, X.; Benyon, R.C. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J. Hepatol. 2002, 36, 362–369. [Google Scholar]
- Knight, V.; Tchongue, J.; Lourensz, D.; Tipping, P.; Sievert, W. Protease-activated receptor 2 promotes experimental liver fibrosis and activates human hepatic stellate cells. Hepatology 2011. [Google Scholar]
- Alves Pedroso, M.L.; Boldt, A.B.; Pereira-Ferrari, L.; Steffensen, R.; Strauss, E.; Jensenius, J.C.; Ioshii, S.O.; Messias-Reason, I. Mannan-binding lectin MBL2 gene polymorphism in chronic hepatitis C: Association with the severity of liver fibrosis and response to interferon therapy. Clin. Exp. Immunol. 2008, 152, 258–264. [Google Scholar]
- El Saadany, S.A.; Ziada, D.H.; Farrag, W.; Hazaa, S. Fibrosis severity and mannan-binding lectin (MBL)/MBL-associated serine protease 1 (MASP-1) complex in HCV-infected patients. Arab. J. Gastroenterol. 2011, 12, 68–73. [Google Scholar]
- Agnello, V.; Chung, R.T.; Kaplan, L.M. A role for hepatitis C virus infection in type II cryoglobulinemia. N. Engl. J. Med. 1992, 327, 1490–1495. [Google Scholar]
- De Re, V.; Sansonno, D.; Simula, M.P.; Caggiari, L.; Gasparotto, D.; Fabris, M.; Tucci, F.A.; Racanelli, V.; Talamini, R.; Campagnolo, M.; et al. HCV-NS3 and IgG-Fc crossreactive IgM in patients with type II mixed cryoglobulinemia and B-cell clonal proliferations. Leukemia 2006, 20, 1145–1154. [Google Scholar] [PubMed]
- Cacoub, P.; Musset, L.; Amoura, Z.; Guilani, P.; Chabre, H.; Lunel, F.; Poynard, T.; Opolon, P.; Piette, J.C. Anticardiolipin, anti-beta2-glycoprotein I, and antinucleosome antibodies in hepatitis C virus infection and mixed cryoglobulinemia. Multivirc group. J. Rheumatol. 1997, 24, 2139–2144. [Google Scholar] [PubMed]
- Lienesch, D.W.; Sherman, K.E.; Metzger, A.; Shen, G.Q. Anti-Clq antibodies in patients with chronic hepatitis C infection. Clin. Exp. Rheumatol. 2006, 24, 183–185. [Google Scholar]
- Sansonno, D.; Tucci, F.A.; Ghebrehiwet, B.; Lauletta, G.; Peerschke, E.I.; Conteduca, V.; Russi, S.; Gatti, P.; Sansonno, L.; Dammacco, F. Role of the receptor for the globular domain of C1q protein in the pathogenesis of hepatitis C virus-related cryoglobulin vascular damage. J. Immunol. 2009, 183, 6013–6020. [Google Scholar]
- Sansonno, D.; Lauletta, G.; Nisi, L.; Gatti, P.; Pesola, F.; Pansini, N.; Dammacco, F. Non-enveloped HCV core protein as constitutive antigen of cold-precipitable immune complexes in type II mixed cryoglobulinaemia. Clin. Exp. Immunol. 2003, 133, 275–282. [Google Scholar]
- Ohsawa, I.; Ohi, H.; Tamano, M.; Endo, M.; Fujita, T.; Satomura, A.; Hidaka, M.; Fuke, Y.; Matsushita, M. Cryoprecipitate of patients with cryoglobulinemic glomerulonephritis contains molecules of the lectin complement pathway. Clin. Immunol. 2001, 101, 59–66. [Google Scholar]
- Rosa, D.; Saletti, G.; De Gregorio, E.; Zorat, F.; Comar, C.; D'Oro, U.; Nuti, S.; Houghton, M.; Barnaba, V.; Pozzato, G.; et al. Activation of naive B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 18544–18549. [Google Scholar] [PubMed]
- Machida, K.; Cheng, K.T.; Pavio, N.; Sung, V.M.; Lai, M.M. Hepatitis C virus E2-CD81 interaction induces hypermutation of the immunoglobulin gene in B cells. J. Virol. 2005, 79, 8079–8089. [Google Scholar]
- Yao, Z.Q.; Eisen-Vandervelde, A.; Ray, S.; Hahn, Y.S. HCV core/gC1qR interaction arrests T cell cycle progression through stabilization of the cell cycle inhibitor p27KIP1. Virology 2003, 314, 271–282. [Google Scholar]
- Yao, Z.Q.; Shata, M.T.; Tricoche, N.; Shan, M.M.; Brotman, B.; Pfahler, W.; Hahn, Y.S.; Prince, A.M. gC1qR expression in chimpanzees with resolved and chronic infection: Potential role of HCV core/gC1qR-mediated T cell suppression in the outcome of HCV infection. Virology 2006, 346, 324–337. [Google Scholar]
- Rickert, R.C. Regulation of B lymphocyte activation by complement C3 and the B cell coreceptor complex. Curr. Opin. Immunol. 2005, 17, 237–243. [Google Scholar]
- Nakayama, Y.; Kim, S.I.; Kim, E.H.; Lambris, J.D.; Sandor, M.; Suresh, M. C3 promotes expansion of CD8+ and CD4+ T cells in a Listeria monocytogenes infection. J. Immunol. 2009, 183, 2921–2931. [Google Scholar]
- Suresh, M.; Molina, H.; Salvato, M.S.; Mastellos, D.; Lambris, J.D.; Sandor, M. Complement component 3 is required for optimal expansion of CD8+ T cells during a systemic viral infection. J. Immunol. 2003, 170, 788–794. [Google Scholar]
- Friedman, H.M.; Wang, L.; Pangburn, M.K.; Lambris, J.D.; Lubinski, J. Novel mechanism of antibody-independent complement neutralization of herpes simplex virus type 1. J. Immunol. 2000, 165, 4528–4536. [Google Scholar]
- Michelow, I.C.; Dong, M.; Mungall, B.A.; Yantosca, L.M.; Lear, C.; Ji, X.; Karpel, M.; Rootes, C.L.; Brudner, M.; Houen, G.; et al. A novel L-ficolin/mannose-binding lectin chimeric molecule with enhanced activity against ebola virus. J. Biol. Chem. 2010, 285, 24729–24739. [Google Scholar] [PubMed]
- Tarr, A.W.; Urbanowicz, R.A.; Hamed, M.R.; Albecka, A.; McClure, C.P.; Brown, R.J.; Irving, W.L.; Dubuisson, J.; Ball, J.K. Hepatitis C patient-derived glycoproteins exhibit marked differences in susceptibility to serum neutralizing antibodies: Genetic subtype defines antigenic but not neutralization serotype. J. Virol. 2011, 85, 4246–4257. [Google Scholar]
- Meuleman, P.; Bukh, J.; Verhoye, L.; Farhoudi, A.; Vanwolleghem, T.; Wang, R.Y.; Desombere, I.; Alter, H.; Purcell, R.H.; Leroux-Roels, G. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 2011, 53, 755–762. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tarr, A.W.; Urbanowicz, R.A.; Ball, J.K. The Role of Humoral Innate Immunity in Hepatitis C Virus Infection. Viruses 2012, 4, 1-27. https://doi.org/10.3390/v4010001
Tarr AW, Urbanowicz RA, Ball JK. The Role of Humoral Innate Immunity in Hepatitis C Virus Infection. Viruses. 2012; 4(1):1-27. https://doi.org/10.3390/v4010001
Chicago/Turabian StyleTarr, Alexander W., Richard A. Urbanowicz, and Jonathan K. Ball. 2012. "The Role of Humoral Innate Immunity in Hepatitis C Virus Infection" Viruses 4, no. 1: 1-27. https://doi.org/10.3390/v4010001
APA StyleTarr, A. W., Urbanowicz, R. A., & Ball, J. K. (2012). The Role of Humoral Innate Immunity in Hepatitis C Virus Infection. Viruses, 4(1), 1-27. https://doi.org/10.3390/v4010001