Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel

Abstract
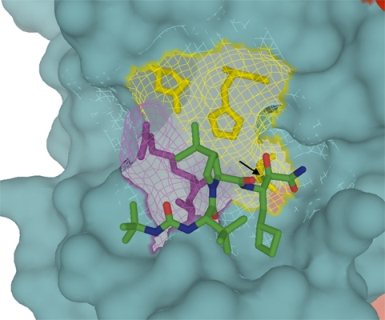
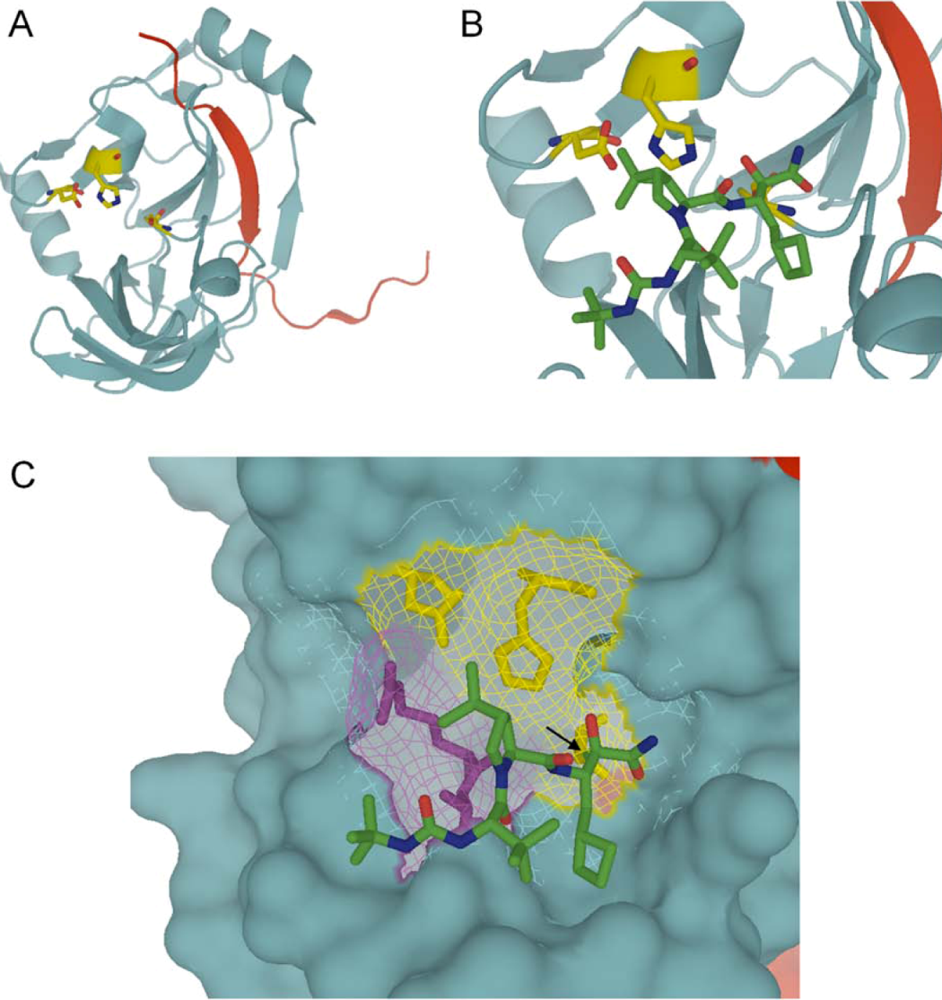
:
1. Introduction
2. Need for improved HCV therapeutic regiments using combination strategies
3. Design of NS3 protease inhibitor BILN 2061: First anti-HCV proof-of-concept in man
4. NS3 protease inhibitors in clinical development
4.1. Telaprevir
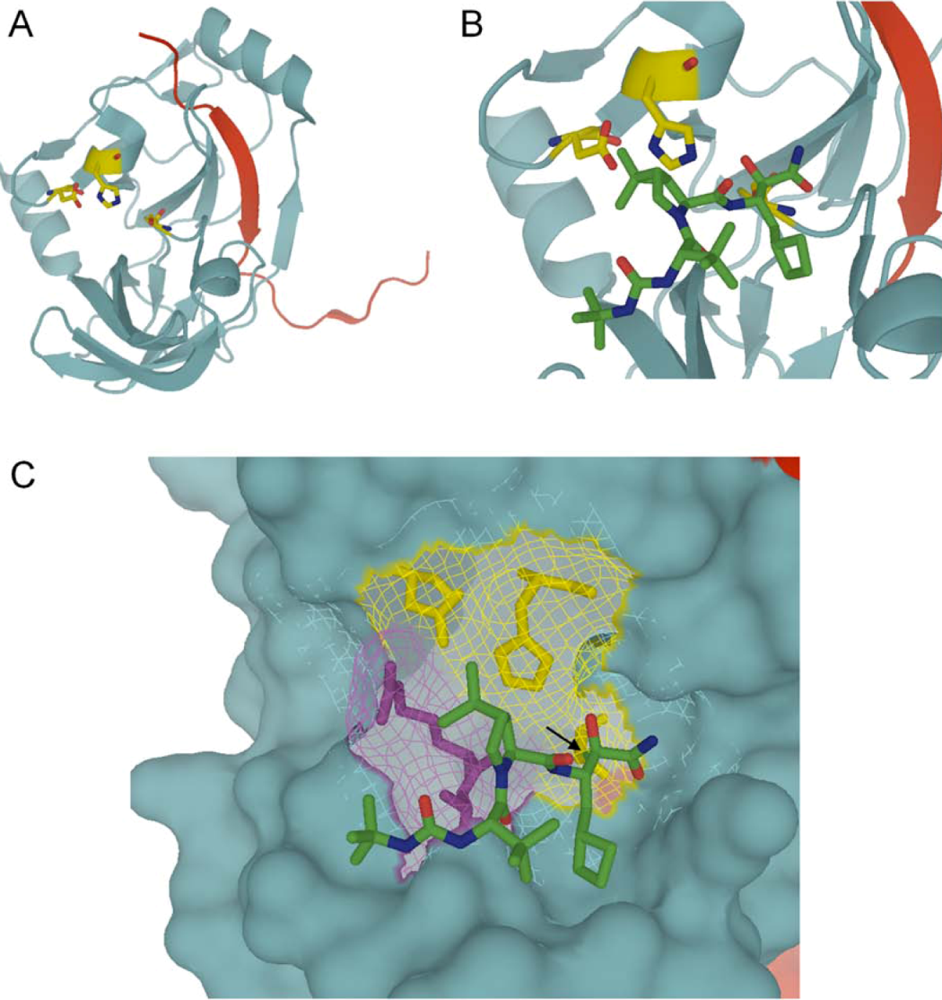

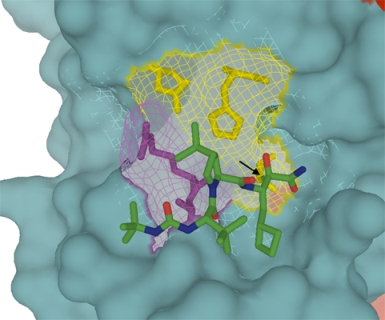{kind=link}
{kind=link}
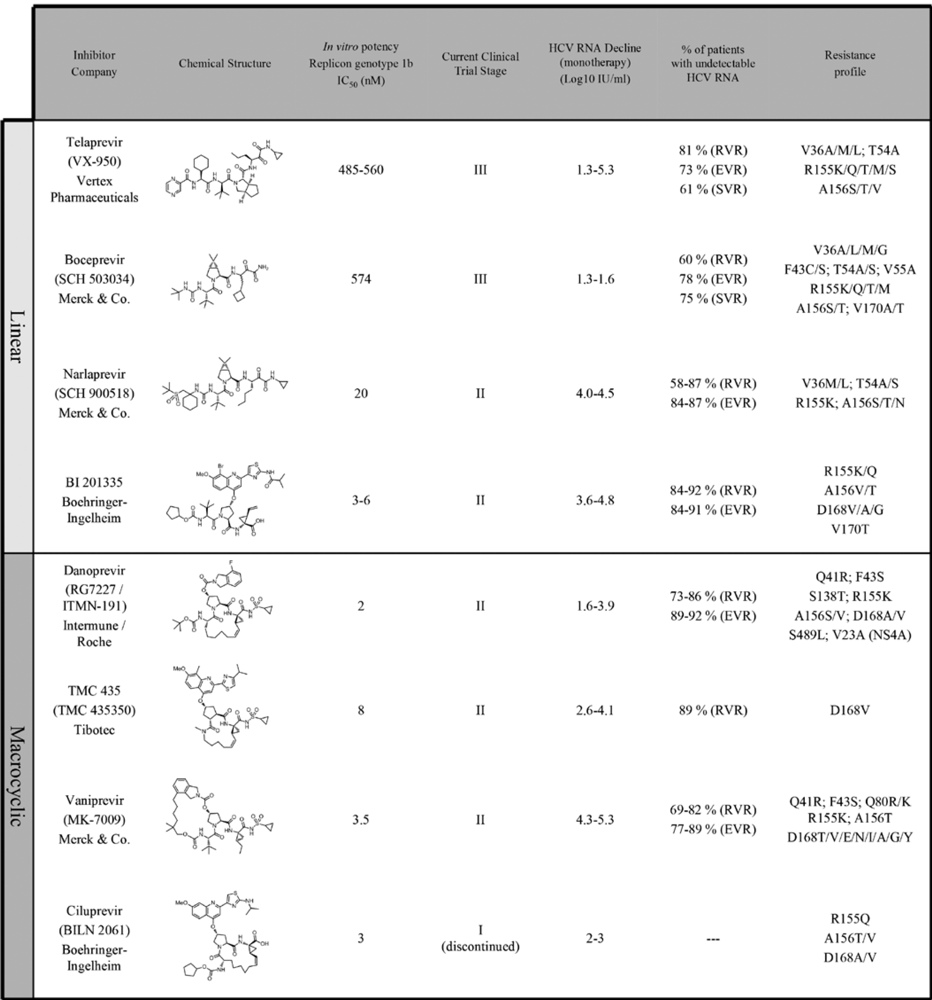 |
4.2. Boceprevir
4.3. TMC 435
4.4. Danoprevir
4.5. Other NS3 protease inhibitors
5. Challenges and future directions
Acknowledgments
References
- Kolykhalov, A.A.; Mihalik, K.; Feinstone, S.M.; Rice, C.M. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3' nontranslated region are essential for virus replication in vivo. J. Virol. 2000, 74, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Dustin, L.B.; Rice, C.M. Flying under the radar: the immunobiology of hepatitis C. Annu. Rev. Immunol. 2007, 25, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Sette Jr., H.; Morgan, T.R.; Balan, V.; Diago, M.; Marcellin, P.; Ramadori, G.; Bodenheimer Jr., H.; Bernstein, D.; Rizzetto, M.; Zeuzem, S.; Pockros, P.J.; Lin, A.; Ackrill, A.M. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [Google Scholar] [PubMed]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; O'Malley, E.T.; Harbeson, S.L.; Rice, C.M.; Murcko, M.A.; Caron, P.R.; Thomson, J.A. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 1996, 87, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Owen, D.M.; Li, K.; Erickson, A.K.; Johnson, C.L.; Fish, P.M.; Carney, D.S.; Wang, T.; Ishida, H.; Yoneyama, M.; Fujita, T.; Saito, T.; Lee, W.M.; Hagedorn, C.H.; Lau, D.T.; Weinman, S.A.; Lemon, S.M.; Gale Jr., M. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6001–6006. [Google Scholar] [CrossRef] [PubMed]
- Lamarre, D.; Anderson, P.C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bos, M.; Cameron, D.R.; Cartier, M.; Cordingley, M.G.; Faucher, A.M.; Goudreau, N.; Kawai, S.H.; Kukolj, G.; Lagace, L.; LaPlante, S.R.; Narjes, H.; Poupart, M.A.; Rancourt, J.; Sentjens, R.E.; St George, R.; Simoneau, B.; Steinmann, G.; Thibeault, D.; Tsantrizos, Y.S.; Weldon, S.M.; Yong, C.L.; Llinas-Brunet, M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Hezode, C.; Forestier, N.; Dusheiko, G.; Ferenci, P.; Pol, S.; Goeser, T.; Bronowicki, J.P.; Bourliere, M.; Gharakhanian, S.; Bengtsson, L.; McNair, L.; George, S.; Kieffer, T.; Kwong, A.; Kauffman, R.S.; Alam, J.; Pawlotsky, J.M.; Zeuzem, S. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 2009, 360, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Everson, G.T.; Gordon, S.C.; Jacobson, I.M.; Sulkowski, M.; Kauffman, R.; McNair, L.; Alam, J.; Muir, A.J. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 2009, 360, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Muir, A.; Adda, N.; Jacobson, I.; Afdhal, N.; Heathcote, J.; Zeuzem, S.; Reesink, H.; Terrault, N.; Bsharat, M.; George, S.; McHutchison, J.G.; Di Bisceglie, A. Telaprevir in hepatitis C genotype 1 infected patients with prior non-response, viral breakthrough or relapse tp peginterferon-alfa-2a/b and ribavirin therapy: SVR results of the PROVE 3 study . J. Hepatol. 2009, 50, S379. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Manns, M.P.; Muir, A.J.; Terrault, N.A.; Jacobson, I.M.; Afdhal, N.H.; Heathcote, E.J.; Zeuzem, S.; Reesink, H.W.; Garg, J.; Bsharat, M.; George, S.; Kauffman, R.S.; Adda, N.; Di Bisceglie, A.M. Telaprevir for previously treated chronic HCV infection. N. Engl. J. Med. 2010, 362, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- 75% of Treatment-Naïve Patients with Chronic Hepatitis C Achieve SVR (Viral Cure) with Telaprevir-Based Treatment in Phase 3 Trial . Available online: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=473342 (accessed on 2 June 2010).
- Kwo, P.; Lawitz, E.J.; McCone, J.; Schiff, E.R.; Vierling, J.M.; Pound, D.; Davis, M.; Galati, J.S.; Gordon, S.C.; Ravendhran, N.; Rossaro, L.; Anderson, F.H.; Jacobson, I.M.; Rubin, R.; Koury, K.; Boparai, N.; Chaudhri, E.; Brass, C.A.; Albrecht, J.K. High sustained virologic response (SVR) in genotype 1 (G1) null responders to PEG-interferon alfa-2b (P) plus ribavirin (R) when treated with boceprevir (BOC) combination therapy . Presented at the AASLD 60th Annual Meeting, Boston, Massachusetts, USA; 29 10 2009. [Google Scholar]
- Schiff, E.R.; Poordad, F.; Jacobson, I.; Flamm, S.; Bacon, B.; Lawitz, E.J.; Gordon, S.C.; McHutchison, J.G.; Ghalib, R.; Poynard, T.; Sulkowski, M.S.; Trepo, C.; Rizzetto, M.; Zeuzem, S.; Marcellin, P.; Mendez, P.; Brass, C.; Albrecht, J.K. Boceprevir (B) combination therapy in null responders (NR): Response dependent on interferon responsiveness . Presented at the EASL 43rd Annual Meeting; 23-27 4 2008. [Google Scholar]
- Marcellin, P.; Reesink, H.; Berg, T.; Cramp, M.; Flisiak, R.; Van Vlierberghe, H.; Verloes, R.; Lenz, O.; Peeters, M.; Sekar, V.; De Smedt, G. Antiviral activity and safety of TMC435 combined with peginterferon alpha-2a and ribavirin in patients with genotype-1 hepatitis C infection who failed previous IFN-based therapy. Presented at the EASL 44th Annual Meeting, Copenhagen, Denmark; 22-26 4 2009. [Google Scholar]
- Forestier, N.; Larrey, D.; Marcellin, P.; Benhamou, Y.; Guyader, D.; Bradford, W.; Porter, S.; Patat, A.; Rouzier, R.; Zeuzem, S. Antiviral activity and safety of ITMN-191 in combination with Peginterferon alfa-2A and ribavirin in patients with chronic hepatitis C virus (HCV) . Presented at the EASL 44th Annual Meeting, Copenhagen, Denmark; 22-26 4 2009. [Google Scholar]
- InterMune (and Roche: phase 2b RG7227/ITMN-191). Reports on Hepatitis C Treatment at EASL . Available online: http://www.natap.org/2010/HCV/041510_01.htm (accessed on 2 June 2010).
- Gane, E.J.; Roberts, S.K.; Stedman, C.; Angus, P.W.; Ritchie, B.; Elston, R.; Ipe, D.; Morcos, P.; Najera, I.; Chu, T.; Berrey, M.M.; Bradford, W.; Laughlin, M.; Shulman, N.S.; Smith, P.S. Combination therapy with a nucleoside polymerase (R7128) and protease (R7227/ITMN-191) inhibitor in HCV: Safety, pharmacokinetics, and virologic results from INFORM-1 . Presented at the AASLD 60th Annual Meeting; 10 2009. [Google Scholar]
- Gane, E.; Roberts, S.; Stedman, C.; Angus, P.; Ritchie, B.; Elston, R.; Ipe, D.; Morcos, P.; Baher, L.; Najera, I.; Chu, T.; Mannino, M.; Berry, M.; Bradford, W.; Laughlin, M.; Shulman, N.; Smith, P. Early On-Teatment Response During Pegylated Interferon Plus Ribavirin are Increased Following 13 Days of Combination Nucleoside Polymerase (RG7128) and Protease (RG7227) Inhibitor Therapy (INFORM-1) . Presented at the EASL 45th Annual Meeting, Vienna, Austria; 4 2010. [Google Scholar]
- Manns, M.P.; Gane, E.; Rodriguez-torres, M.; Stoehr, A.; Yeh, C.T.; Marcellin, P.; Wiedmann, R.T.; Hwang, P.M.; Barnard, R.J.; Quirk, E.; Kartsonis, N.A.; Lee, A.W. Early viral response (EVR) rates in treatment-naïve patients with chronic hepatitis C (CHC) genotype 1 infection treated with MK-7009, a novel NS3/4A protease inhibitor, in combination with pegylated interferon alfa-2a and ribavirin for 28 days . Presented at the AASLD 60th Annual Meeting, Boston, Massachusetts, USA; 29 10 2009. [Google Scholar]
- Vierling, J.M.; Poordad, F.; Lawitz, E.J.; Ghalib, R.H.; Lee, W.M.; Ravendhran, N.; Galati, J.S.; Bacon, B.R.; Flamm, S.L.; Balart, L.A.; Freilich, B.; Schiff, E.R.; Jacobson, I.M.; Kwo, P.Y.; Gordon, S.C.; Sulkowski, M.S.; Boparai, N.; Chaudhri, E.I.; Brass, C.; Hughes, E.A.; Albrecht, J.K. Once daily Narlaprevir (SCH 900518) in combination with PEGINTRONTM (peginterferon alfa-2b)/ribavirin for teatment-naïve subjects with genotype-1 CHC: Interim results from NEXT-1, a phase 22 study . Presented at the AASLD 60th Annual Meeting, Boston, Massachusetts, USA; 29 10 2009. [Google Scholar]
- Sulkowski, M.S.; Ferenci, P.; Emanoil, C.; Asselah, T.; Caruntu, F.; Lalezari, J.; Bourlière, M.; Mauss, S.; Grange, J.D.; Berg, T.; Zeuzem, S.; Streinu-Cercel, A.; Wright, D.; Jensen, D.M.; Haefner, C.; Datsenko, Y.; Stern, J.O.; Nehmiz, G.; Steinmann, G. SILEN-C1: Early antiviral activity and safety of BI 201335 combined with peginterferon alfa-2a and ribavirin in treatment-naïve patients with chronic genotype 1 HCV infection . Presented at the AASLD 60th Annual Meeting, Boston, Massachusetts, USA; 29 10 2009. [Google Scholar]
- Sulkowski, M.; Bourliere, M.; Bronowicki, J.P.; Streinu-Cercel, A.; Preotescu, L.; Asselah, T.; Pawlotsky, J.M.; Shafran, S.; Pol, S.; Caruntu, F.A.; Mauss, S.; Larrey, D.; Häfner, C.; Datsenko, Y.; Stern, J.O.; Kubiak, R.; Steinmann, G. SILEN-C2: Early antiviral activity and safety of BI 201335 combined with peginterferon alfa-2a and ribavirin (PegIFN/RBV) in chronic HCV genotype 1 patients with non-response to PegIFN/RBV . Presented at the EASL 45th Annual Meeting, Vienna, Austria; 4 2010. [Google Scholar]
- Foster, G.R.; Hezode, C.; Bronowicki, J.P.; Carosi, G.; Weiland, O.; Verlinden, L.; Van Heeswijk, R.; Van Baelen, B.; Picchio, G.; Beumont-Mauviel, M. Activity of Telaprevir Alone or in Combination with Peginterferon Alfa-2a and Ribavirin in Treatment-naïve, Genotype 2 and 3, Hepatitis C Patients: Final Results of Study C209 . Presented at the EASL 45th Annual Meeting, Vienna, Austria; 4 2010. [Google Scholar]
- Liverton, L. MK-5172, the 1st HCV Protease Inibitor with Potent Acyivity Against Resistance Mutations In Vitro. Presented at the EASL 45th Annual Meeting, Vienna, Austria; 4 2010. [Google Scholar]
- Beran, R.K.; Pyle, A.M. Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J. Biol. Chem. 2008, 283, 29929–29937. [Google Scholar] [CrossRef] [PubMed]
- Beran, R.K.; Serebrov, V.; Pyle, A.M. The serine protease domain of hepatitis C viral NS3 activates RNA helicase activity by promoting the binding of RNA substrate. J. Biol. Chem. 2007, 282, 34913–34920. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, V.; Gurjar, M.; Levin, M.K.; Patel, S.S. The Protease Domain Increases the Translocation Stepping Efficiency of the Hepatitis C Virus NS3-4A Helicase. J. Biol. Chem. 2010, 285, 17821–17832. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; Riordan, S.; Sheridan, D.; Smedile, A.; Fragomeli, V.; Muller, T.; Bahlo, M.; Stewart, G.J.; Booth, D.R.; George, J. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Akuta, N.; Suzuki, F.; Hirakawa, M.; Kawamura, Y.; Yatsuji, H.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Arase, Y.; Ikeda, K.; Chayama, K.; Nakamura, Y.; Kumada, H. Amino acid substitution in hepatitis C virus core region and genetic variation near the interleukin 28B gene predict viral response to telaprevir with peginterferon and ribavirin. Hepatology 2010, 52, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Prongay, A.J.; Guo, Z.; Yao, N.; Pichardo, J.; Fischmann, T.; Strickland, C.; Myers Jr., J.; Weber, P.C.; Beyer, B.M.; Ingram, R.; Hong, Z.; Prosise, W.W.; Ramanathan, L.; Taremi, S.S.; Yarosh-Tomaine, T.; Zhang, R.; Senior, M.; Yang, R.S.; Malcolm, B.; Arasappan, A.; Bennett, F.; Bogen, S.L.; Chen, K.; Jao, E.; Liu, Y.T.; Lovey, R.G.; Saksena, A.K.; Venkatraman, S.; Girijavallabhan, V.; Njoroge, F.G.; Madison, V. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3- [2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl] - 6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem. 2007, 50, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Chatel-Chaix, L.; Baril, M.; Lamarre, D. Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel. Viruses 2010, 2, 1752-1765. https://doi.org/10.3390/v2081752
Chatel-Chaix L, Baril M, Lamarre D. Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel. Viruses. 2010; 2(8):1752-1765. https://doi.org/10.3390/v2081752
Chicago/Turabian StyleChatel-Chaix, Laurent, Martin Baril, and Daniel Lamarre. 2010. "Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel" Viruses 2, no. 8: 1752-1765. https://doi.org/10.3390/v2081752
APA StyleChatel-Chaix, L., Baril, M., & Lamarre, D. (2010). Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel. Viruses, 2(8), 1752-1765. https://doi.org/10.3390/v2081752