Last Stop Before Exit – Hepatitis C Assembly and Release as Antiviral Drug Targets

{kind=link}
{kind=link}
Abstract
:1. Introduction
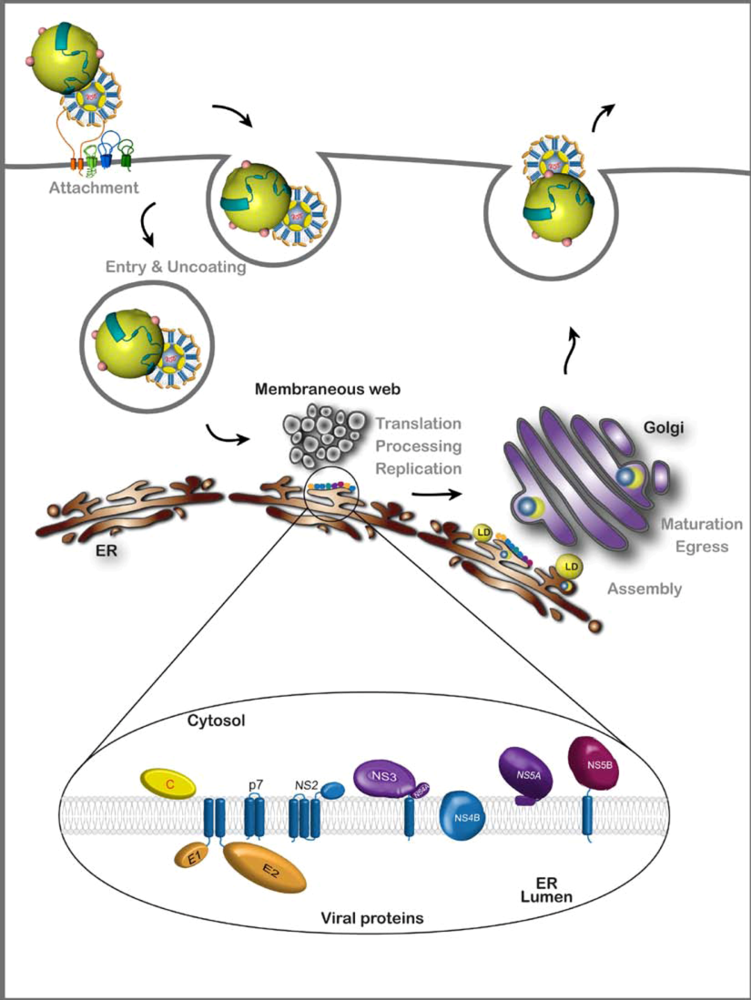
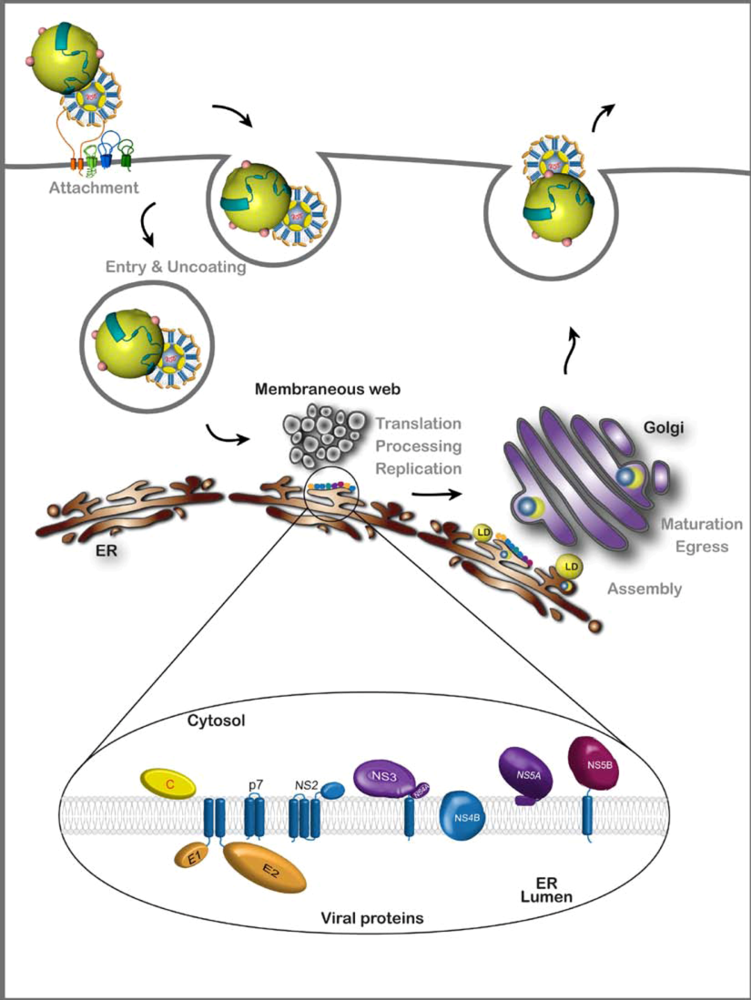
2. HCV Assembly
2.1. Viral components of the assembly machinery
2.3. Involvement of p7 and NS2 in HCV assembly
2.4. HCV envelope glycoproteins and virion assembly
2.5. Very low density lipoproteins (VLDL) and HCV assembly
2.6. Viral particle transport and release
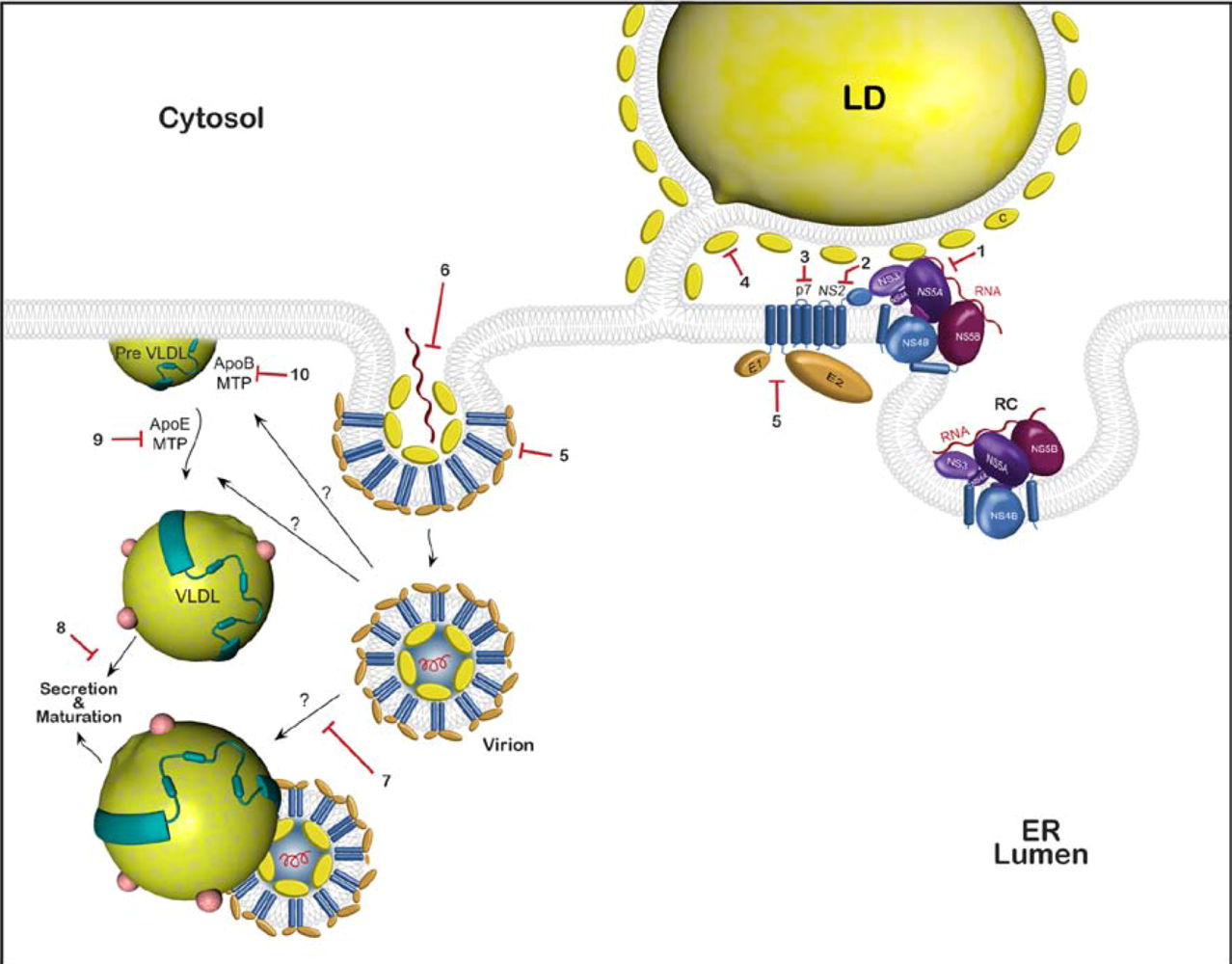
3. Drug targets
3.1. Possible viral targets and existing drugs
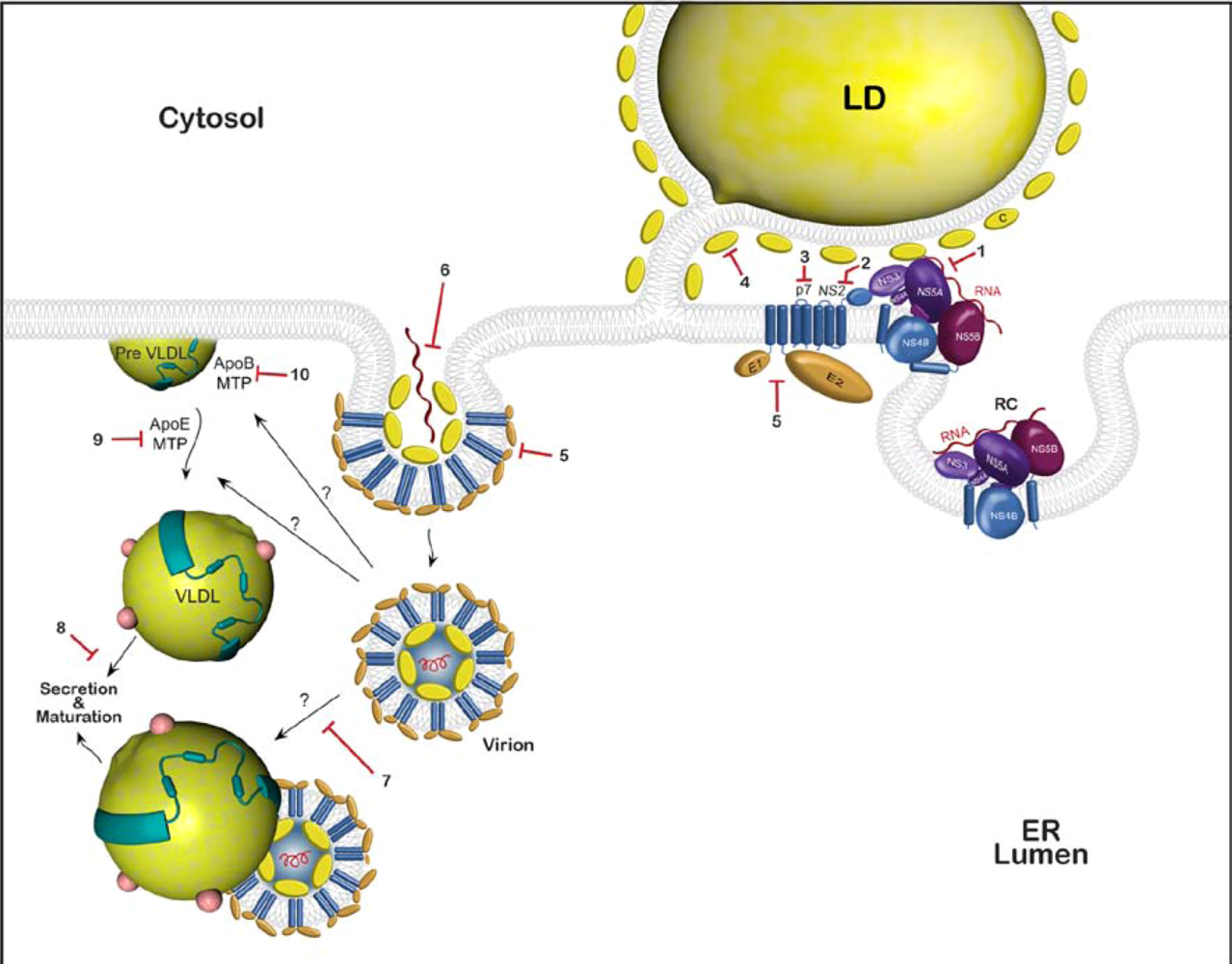
3.2. Possible cellular targets
3.3. Approved drugs that might potentially inhibit HCV assembly
4. Conclusions
Acknowledgments
References
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; Walker, C.; Alter, M.J.; Yi, M. Hepatitis C Virus, 5th ed; Lippincott Williams and Wilkins: Philadelphia, PA, U. S. A., 2007. [Google Scholar]
- Varaklioti, A.; Vassilaki, N.; Georgopoulou, U.; Mavromara, P. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J. Biol. Chem. 2002, 277, 17713–17721. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Scheel, T.K.; Jensen, T.B.; Lademann, J.B.; Prentoe, J.C.; Knudsen, M.L.; Hoegh, A.M.; Bukh, J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 2009, 49, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Burlone, M.E.; Budkowska, A. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J. Gen. Virol. 2009, 90, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouillé, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Dragic, T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 2006, 80, 11571–11578. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Gosert, R.; Egger, D.; Penin, F.; Blum, H.E.; Bienz, K. Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antiviral. Res. 2003, 60, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient initiation of HCV RNA replication in cell culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Micro. 2008, 6, 699–708. [Google Scholar] [CrossRef]
- Barba, G.; Harper, F.; Harada, T.; Kohara, M.; Goulinet, S.; Matsuura, Y.; Eder, G.; Schaff, Z.; Chapman, M.J.; Miyamura, T.; Brechot, C. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Englert, C.; Wakita, T.; Wands, J.R. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology 1996, 222, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Parton, R.G. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell. Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell. Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Santolini, E.; Migliaccio, G.; La Monica, N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 1994, 68, 3631–3641. [Google Scholar] [PubMed]
- Hussy, P.; Langen, H.; Mous, J.; Jacobsen, H. Hepatitis C virus core protein: carboxy-terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. Virology 1996, 224, 93–104. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Vanbelle, C.; Ebel, C.; Penin, F.; Lavergne, J.P. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J. Virol. 2005, 79, 11353–11365. [Google Scholar] [CrossRef] [PubMed]
- Hope, R.G.; Murphy, D.J.; McLauchlan, J. The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 2002, 277, 4261–4270. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J. Properties of the hepatitis C virus core protein: a structural protein that modulates cellular processes. J. Viral. Hepat. 2000, 7, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Cristofari, G.; Ivanyi-Nagy, R.; Gabus, C.; Boulant, S.; Lavergne, J.P.; Penin, F.; Darlix, J.L. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004, 32, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Shimoike, T.; Mimori, S.; Tani, H.; Matsuura, Y.; Miyamura, T. Interaction of hepatitis C virus core protein with viral sense RNA and suppression of its translation. J. Virol. 1999, 73, 9718–9725. [Google Scholar] [PubMed]
- Nolandt, O.; Kern, V.; Muller, H.; Pfaff, E.; Theilmann, L.; Welker, R.; Krausslich, H.G. Analysis of hepatitis C virus core protein interaction domains . J. Gen. Virol. 1997, 78 ( Pt 6), 1331–1340. [Google Scholar] [PubMed]
- Boulant, S.; Montserret, R.; Hope, R.G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.P.; Penin, F.; McLauchlan, J. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Targett-Adams, P.; McLauchlan, J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 2007, 88, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; Suzuki, T. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef]
- Hanoulle, X.; Badillo, A.; Verdegem, D.; Penin, F.; Lippens, G. The Domain 2 of the HCV NS5A Protein is Intrinsically Unstructured. Protein Pept. Lett. 2010, 17, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Brass, V.; Bieck, E.; Montserret, R.; Wölk, B.; Hellings, J.A.; Blum, H.E.; Penin, F.; Moradpour, D. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2002, 277, 8130–8139. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Brass, V.; Appel, N.; Ramboarina, S.; Montserret, R.; Ficheux, D.; Blum, H.E.; Bartenschlager, R.; Moradpour, D. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [Google Scholar] [CrossRef] [PubMed]
- Hanoulle, X.; Verdegem, D.; Badillo, A.; Wieruszeski, J.-M.; Penin, F.; Lippens, G. Domain 3 of non-structural protein 5A from hepatitis C virus is natively unfolded. Biochem. Biophys. Res. Commun. 2009, 381, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Zayas, M.; Meuleman, P.; Long, G.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Leroux-Roels, G.; Lohmann, V.; Bartenschlager, R. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 2009, 5, e1000475. [Google Scholar] [CrossRef]
- Bukh, J.; Pietschmann, T.; Lohmann, V.; Krieger, N.; Faulk, K.; Engle, R.E.; Govindarajan, S.; Shapiro, M.; St Claire, M.; Bartenschlager, R. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14416–14421. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Rice, C.M.; Goff, S.P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13038–13043. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; McKeating, J.A.; Pietschmann, T.; Frick, D.N.; Glenn, J.S.; Tellinghuisen, T.L.; Symons, J.; Furman, P.A. Development of novel therapies for hepatitis C. Antiviral Res. 2010, 86, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; Suzuki, T. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 2007, 81, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.L.; Jones, C.T.; Tassello, J.; Rice, C.M. Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J. Virol. 2007, 81, 10220–10231. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Beran, R.; Peters, C.; Lorenz, I.; Lindenbach, B. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-4A enzyme complexes. J. Virol. 2009, 83, 8379–95. [Google Scholar] [CrossRef] [PubMed]
- Jirasko, V.; Montserret, R.; Appel, N.; Janvier, A.; Eustachi, L.; Brohm, C.; Steinmann, E.; Pietschmann, T.; Penin, F.; Bartenschlager, R. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 2008, 283, 28546–28562. [Google Scholar] [CrossRef] [PubMed]
- Dentzer, T.G.; Lorenz, I.C.; Evans, M.J.; Rice, C.M. Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J. Virol. 2009, 83, 12702–12713. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Brohm, C.; Steinmann, E.; Friesland, M.; Lorenz, I.C.; Patel, A.; Penin, F.; Bartenschlager, R.; Pietschmann, T. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J. Virol. 2009, 83, 11682–11693. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 2009, 5, e1000403. [Google Scholar] [CrossRef]
- Carrere-Kremer, S.; Montpellier-Pala, C.; Cocquerel, L.; Wychowski, C.; Penin, F.; Dubuisson, J. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 2002, 76, 3720–3730. [Google Scholar] [CrossRef] [PubMed]
- Luik, P.; Chew, C.; Aittoniemi, J.; Chang, J.; Wentworth, P.; Dwek, R.; Biggin, P.; Vénien-Bryan, C.; Zitzmann, N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12712–6. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.D.; Beales, L.P.; Clarke, D.S.; Worsfold, O.; Evans, S.D.; Jaeger, J.; Harris, M.P.; Rowlands, D.J. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003, 535, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, D.; Neville, D.C.; Argaud, O.; Blumberg, B.; Dwek, R.A.; Fischer, W.B.; Zitzmann, N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6104–6108. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, A.; Wilson, L.; Ewart, G.D.; Gage, P.W. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 2004, 557, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.D.; Harvey, R.; Clarke, D.S.; Barclay, W.S.; Harris, M.; Rowlands, D.J. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol. 2004, 85, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Griffin, S.; Beales, L.; Gelais, C.S.; Burgess, S.; Harris, M.; Rowlands, D. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 2006, 281, 37057–37068. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7271–7276. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Goffard, A.; Dubuisson, J. Glycosylation of hepatitis C virus envelope proteins. Biochimie 2003, 85, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Krey, T.; d'Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R.G.; Baron, B.; Robert, B.; England, P.; Persson, M.A.A.; Martin, A.; Rey, F.A. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010, 6, e1000762. [Google Scholar] [CrossRef]
- Cocquerel, L.; Duvet, S.; Meunier, J.C.; Pillez, A.; Cacan, R.; Wychowski, C.; Dubuisson, J. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 1999, 73, 2641–2649. [Google Scholar] [PubMed]
- Cocquerel, L.; Meunier, J.C.; Pillez, A.; Wychowski, C.; Dubuisson, J. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 1998, 72, 2183–2191. [Google Scholar] [PubMed]
- Rouille, Y.; Helle, F.; Delgrange, D.; Roingeard, P.; Voisset, C.; Blanchard, E.; Belouzard, S.; McKeating, J.; Patel, A.H.; Maertens, G.; Wakita, T.; Wychowski, C.; Dubuisson, J. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 2006, 80, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Lavie, M.; Goffard, A.; Dubuisson, J. Assembly of a functional HCV glycoprotein heterodimer. Curr. Issues Mol. Biol. 2007, 9, 71–86. [Google Scholar] [PubMed]
- Deleersnyder, V.; Pillez, A.; Wychowski, C.; Blight, K.; Xu, J.; Hahn, Y.S.; Rice, C.M.; Dubuisson, J. Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 1997, 71, 697–704. [Google Scholar] [PubMed]
- Op De Beeck, A.; Voisset, C.; Bartosch, B.; Ciczora, Y.; Cocquerel, L.; Keck, Z.; Foung, S.; Cosset, F.L.; Dubuisson, J. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 2004, 78, 2994–3002. [Google Scholar] [CrossRef] [PubMed]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the Envelope Glycoproteins Associated with Infectious Hepatitis C Virus . J. Virol. 2010, in press. [Google Scholar]
- Icard, V.; Diaz, O.; Scholtes, C.; Perrin-Cocon, L.; Ramière, C.; Bartenschlager, R.; Penin, F.; Lotteau, V.; André, P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLoS ONE 2009, 4, e4233. [Google Scholar] [CrossRef]
- Pecheur, E.I.; Diaz, O.; Molle, J.; Icard, V.; Bonnafous, P.; Lambert, O.; Andre, P. Morphological characterization and fusion properties of triglyceride-rich lipoproteins obtained from cells transduced with hepatitis C virus glycoproteins. J. Biol. Chem. 2010, 285, 25802–11. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; Syder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; Rice, C.M. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Yao, Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr. Metab. (Lond.) 2010, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale Jr., M.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5848–5853. [Google Scholar] [CrossRef] [PubMed]
- Nahmias, Y.; Goldwasser, J.; Casali, M.; van Poll, D.; Wakita, T.; Chung, R.T.; Yarmush, M.L. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 2008, 47, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Luo, G. Apolipoprotein E but Not B Is Required for the Formation of infectious Hepatitis C Virus Particles. J. Virol. 2009, 83, 12680–91. [Google Scholar] [CrossRef] [PubMed]
- Benga, W.J.A.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; McLauchlan, J.; Baumert, T.F.; Schuster, C. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 2010, 51, 43–53. [Google Scholar] [PubMed]
- Sánchez-San Martín, C.; Liu, C.Y.; Kielian, M. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009, 17, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Kapadia, S.B.; Chisari, F.V. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 2006, 80, 11074–11081. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Buckwold, V.E.; Beer, B.E.; Donis, R.O. Bovine viral diarrhea virus as a surrogate model of hepatitis C virus for the evaluation of antiviral agents. Antiviral Res. 2003, 60, 1–15. [Google Scholar] [CrossRef]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E.; Maerz, A.; Poumbourios, P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003, 546, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Libbrecht, L.; De Vos, R.; de Hemptinne, B.; Gevaert, K.; Vandekerckhove, J.; Roskams, T.; Leroux-Roels, G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 2005, 41, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.; Tyrrell, D.L.; Kneteman, N.M. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001, 7, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Barth, H.; Schuster, C.; Baumert, T.F. Hepatitis C virus entry: molecular mechanisms and targets for antiviral therapy. Front. Biosci. 2009, 14, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; McHutchison, J.G. Antiviral resistance and specifically targeted therapy for HCV (STAT-C). J. Viral. Hepat. 2009, 16, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter Jr., R.; Ikeda, M.; Lemon, S.M.; Gale Jr., M. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Salzwedel, K.; Martin, D.E.; Sakalian, M. Maturation inhibitors: a new therapeutic class targets the virus structure. AIDS Rev. 2007, 9, 162–172. [Google Scholar] [PubMed]
- Flajolet, M.; Rotondo, G.; Daviet, L.; Bergametti, F.; Inchauspe, G.; Tiollais, P.; Transy, C.; Legrain, P. A genomic approach of the hepatitis C virus generates a protein interaction map. Gene 2000, 242, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.; Scampavia, L.; Spicer, T.; Beeler, A.B.; Takahashi, V.; Snyder, J.K.; Porco, J.A.; Hodder, P.; Strosberg, A.D. A time-resolved fluorescence-resonance energy transfer assay for identifying inhibitors of hepatitis C virus core dimerization. Assay Drug Dev. Technol. 2010, 8, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.; Stgelais, C.; Owsianka, A.M.; Patel, A.H.; Rowlands, D.; Harris, M. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 2008, 48, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues. Antiviral Res. 2010, 86, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Whitfield, T.; Kallis, S.; Dwek, R.A.; Zitzmann, N.; Pietschmann, T.; Bartenschlager, R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 2007, 46, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Branza-Nichita, N.; Carrouee-Durantel, S.; Butters, T.D.; Dwek, R.A.; Zitzmann, N. Study of the mechanism of antiviral action of iminosugar derivatives against bovine viral diarrhea virus. J. Virol. 2001, 75, 8987–8998. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R. The NS3/4A proteinase of the hepatitis C virus: unravelling structure and function of an unusual enzyme and a prime target for antiviral therapy. J. Viral. Hepat. 1999, 6, 165–181. [Google Scholar] [CrossRef]
- Kim, D.W.; Gwack, Y.; Han, J.H.; Choe, J. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem. Biophys. Res. Commun. 1995, 215, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O'Boyle, D.R.; Lemm, J.A.; Wang, C.; Knipe, J.O.; Chien, C.; Colonno, R.J.; Grasela, D.M.; Meanwell, N.A.; Hamann, L.G. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect . Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef] [PubMed]
- Branza-Nichita, N.; Petrescu, A.J.; Negroiu, G.; Dwek, R.A.; Petrescu, S.M. N-glycosylation processing and glycoprotein folding-lessons from the tyrosinase-related proteins. Chem. Rev. 2000, 100, 4697–4712. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A.; Butters, T.D.; Platt, F.M.; Zitzmann, N. Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug. Discov. 2002, 1, 65–75. [Google Scholar] [CrossRef]
- Branza-Nichita, N.; Durantel, D.; Carrouee-Durantel, S.; Dwek, R.A.; Zitzmann, N. Antiviral effect of N-butyldeoxynojirimycin against bovine viral diarrhea virus correlates with misfolding of E2 envelope proteins and impairment of their association into E1-E2 heterodimers. J. Virol. 2001, 75, 3527–3536. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.F.; Lee, C.J.; Liao, C.L.; Dwek, R.A.; Zitzmann, N.; Lin, Y.L. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 2002, 76, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, N.; Mehta, A.S.; Carrouée, S.; Butters, T.D.; Platt, F.M.; McCauley, J.; Blumberg, B.S.; Dwek, R.A.; Block, T.M. Imino sugars inhibit the formation and secretion of bovine viral diarrhea virus, a pestivirus model of hepatitis C virus: implications for the development of broad spectrum anti-hepatitis virus agents. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 11878–11882. [Google Scholar] [CrossRef] [PubMed]
- Takkenberg, B.; de Bruijne, J.; Weegink, C.; Jansen, P.; Reesink, H. Novel therapies in hepatitis B and C. Curr. Gastroenterol. Rep. 2008, 10, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M.; Chevaliez, S.; McHutchison, J.G. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007, 132, 1979–1998. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; Rader, D.J. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; McKenney, J.; Bloedon, L.T.; Sasiela, W.J.; Rader, D.J. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, N.; Ishizaka, Y.; Takahashi, E.; Tooda, E.; Hashimoto, H.; Nagai, R.; Yamakado, M. Association between hepatitis C virus seropositivity, carotid-artery plaque, and intima-media thickening. Lancet 2002, 359, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, S.; Bowman, D.; Hussain, M.M.; Alberti, A. Hepatic steatosis in hepatitis C is a storage disease due to HCV interaction with microsomal triglyceride transfer protein (MTP). Nutr. Metab. (Lond.) 2010, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Whitten-Bauer, C.; Chisari, F.V. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, K.; Simeon, R.L.; Rice, C.M.; Chen, Z. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3764–3769. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Tews, B.A.; Popescu, C.-I.; Dubuisson, J. Last Stop Before Exit – Hepatitis C Assembly and Release as Antiviral Drug Targets. Viruses 2010, 2, 1782-1803. https://doi.org/10.3390/v2081782
Tews BA, Popescu C-I, Dubuisson J. Last Stop Before Exit – Hepatitis C Assembly and Release as Antiviral Drug Targets. Viruses. 2010; 2(8):1782-1803. https://doi.org/10.3390/v2081782
Chicago/Turabian StyleTews, Birke Andrea, Costin-Ioan Popescu, and Jean Dubuisson. 2010. "Last Stop Before Exit – Hepatitis C Assembly and Release as Antiviral Drug Targets" Viruses 2, no. 8: 1782-1803. https://doi.org/10.3390/v2081782
APA StyleTews, B. A., Popescu, C.-I., & Dubuisson, J. (2010). Last Stop Before Exit – Hepatitis C Assembly and Release as Antiviral Drug Targets. Viruses, 2(8), 1782-1803. https://doi.org/10.3390/v2081782