Core as a Novel Viral Target for Hepatitis C Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Core as a target protein for anti-HCV drug discovery
3. Core’s role in HCV’s life cycle
3.1. Core interactions with other HCV proteins
3.2. Core’s role in assembly

4. Core’s structure and function
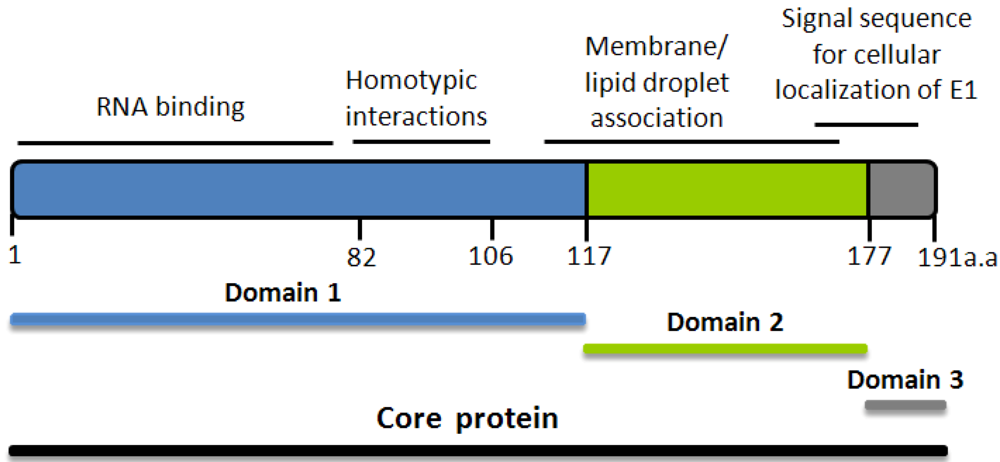
5. Assays for analyzing core function:
a. Assays for Nucleocapsid formation
b. Core dimerization
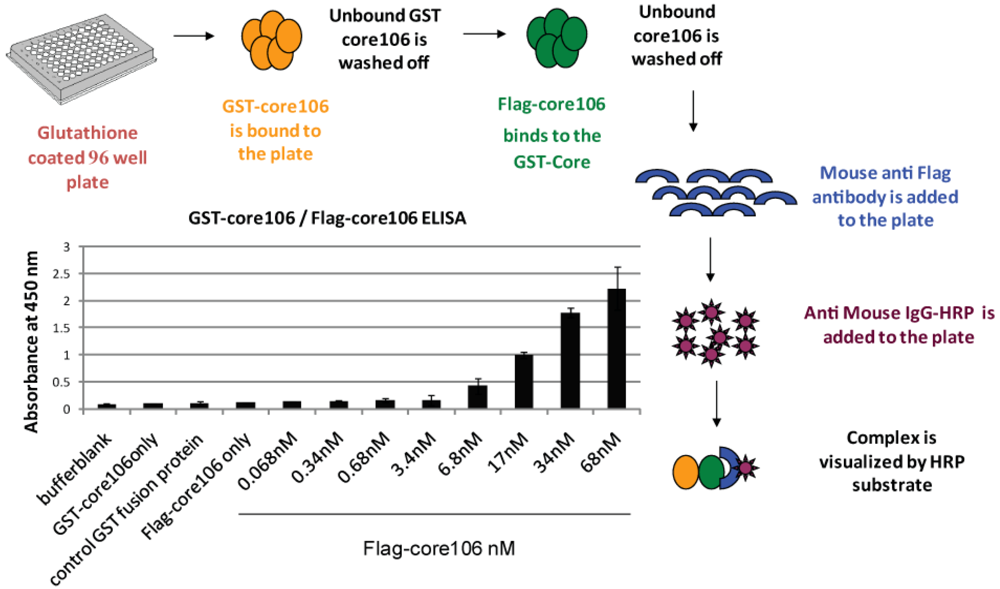
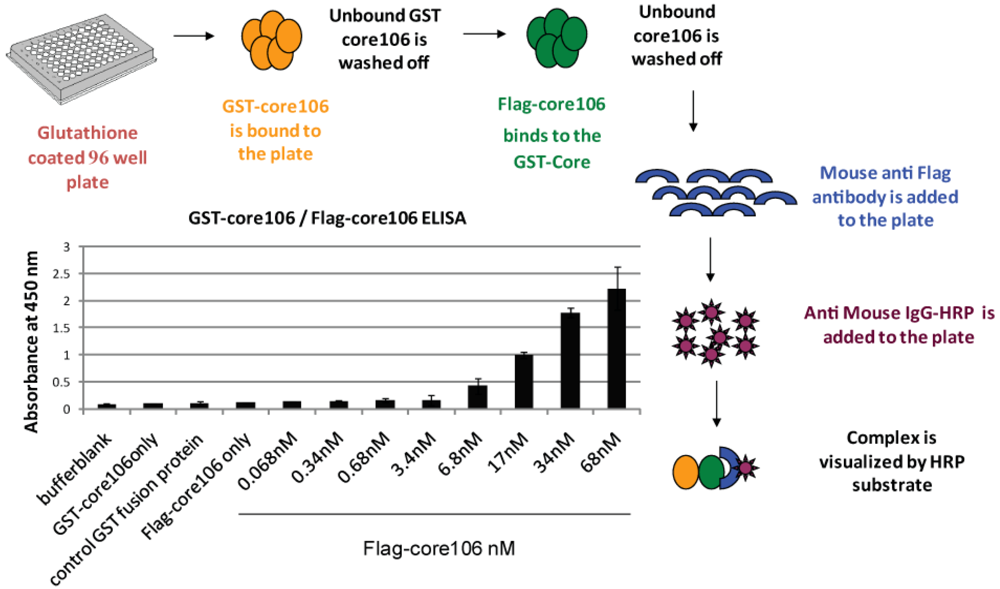
- 1. For developing a sandwich ELISA, GST-tagged core106 domain was adsorbed on microplates coated with glutathione, and a horse-radish peroxidase goat anti-mouse antibody was used to demonstrate binding of anti-Flag antibody to Flag-tagged core106 itself bound to GST-core106 (Figure 3). This result demonstrated hetero-dimerization of the two fusion proteins. Free GST, Flag peptide or untagged core106 each displaced specific binding to background levels. [23].
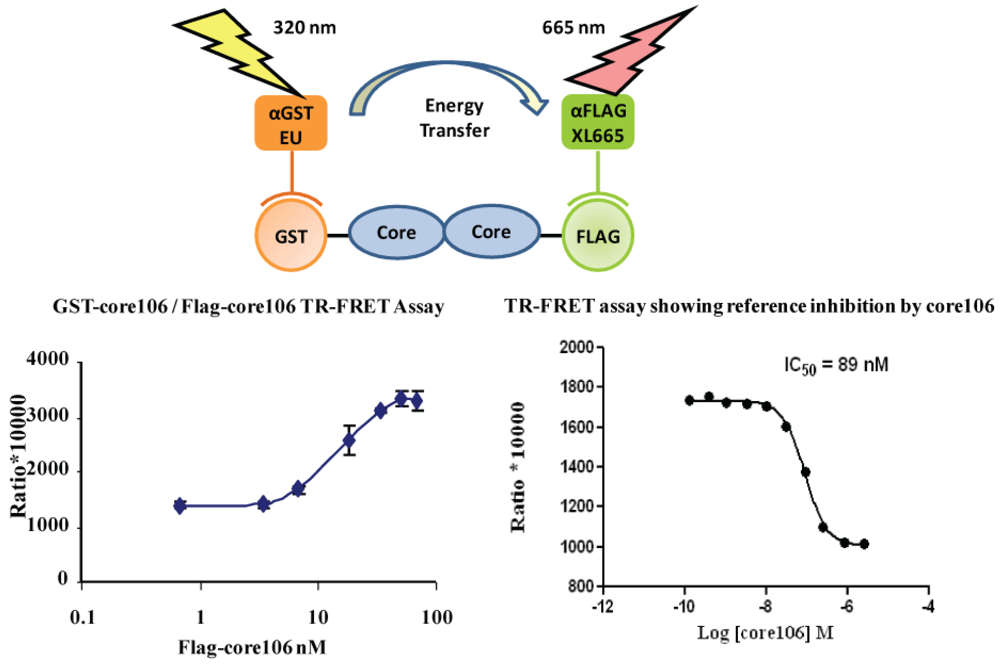
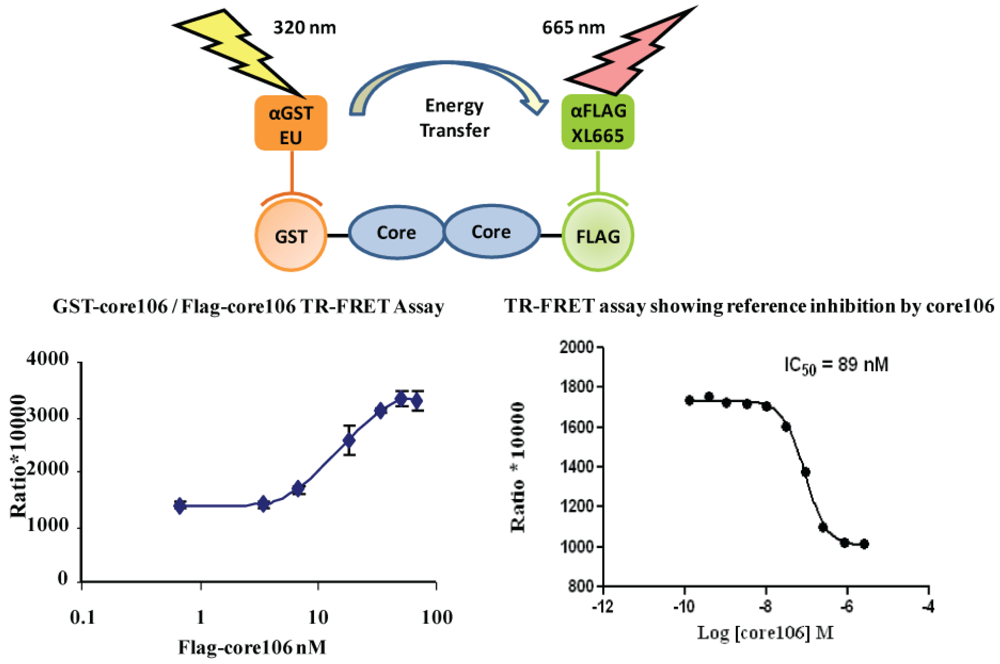
- 2. The ELISA was useful to qualitatively demonstrate the interaction when using alternately tagged core106 fusion proteins, but the need for multiple washes hampered a precise analysis of the interaction, and precluded its use for screening of large compound libraries. For this purpose, a TR-FRET homogenous assay was developed, using the same GST-and Flag-tagged core106 proteins (Figure 4) [23,69]. Fluorophore-labeled antibodies against the GST and Flag tags allowed the authors to quantitatively measure the formation of dimers and oligomers of core106. The TR-FRET was particularly well adapted for robotized high-throughput screening in 384- and 1,536-well microplates of large small compound libraries [69]. The typical signal to background ratio shown in Figure 4 left, was rather low, although the inhibitory signal (Figure 4 right) was quite adequate for identifying “primary hit” inhibitors [69].
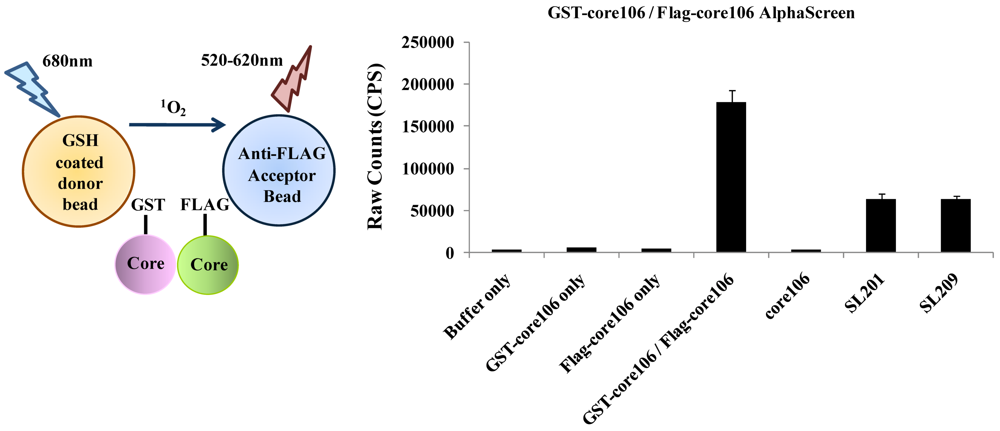
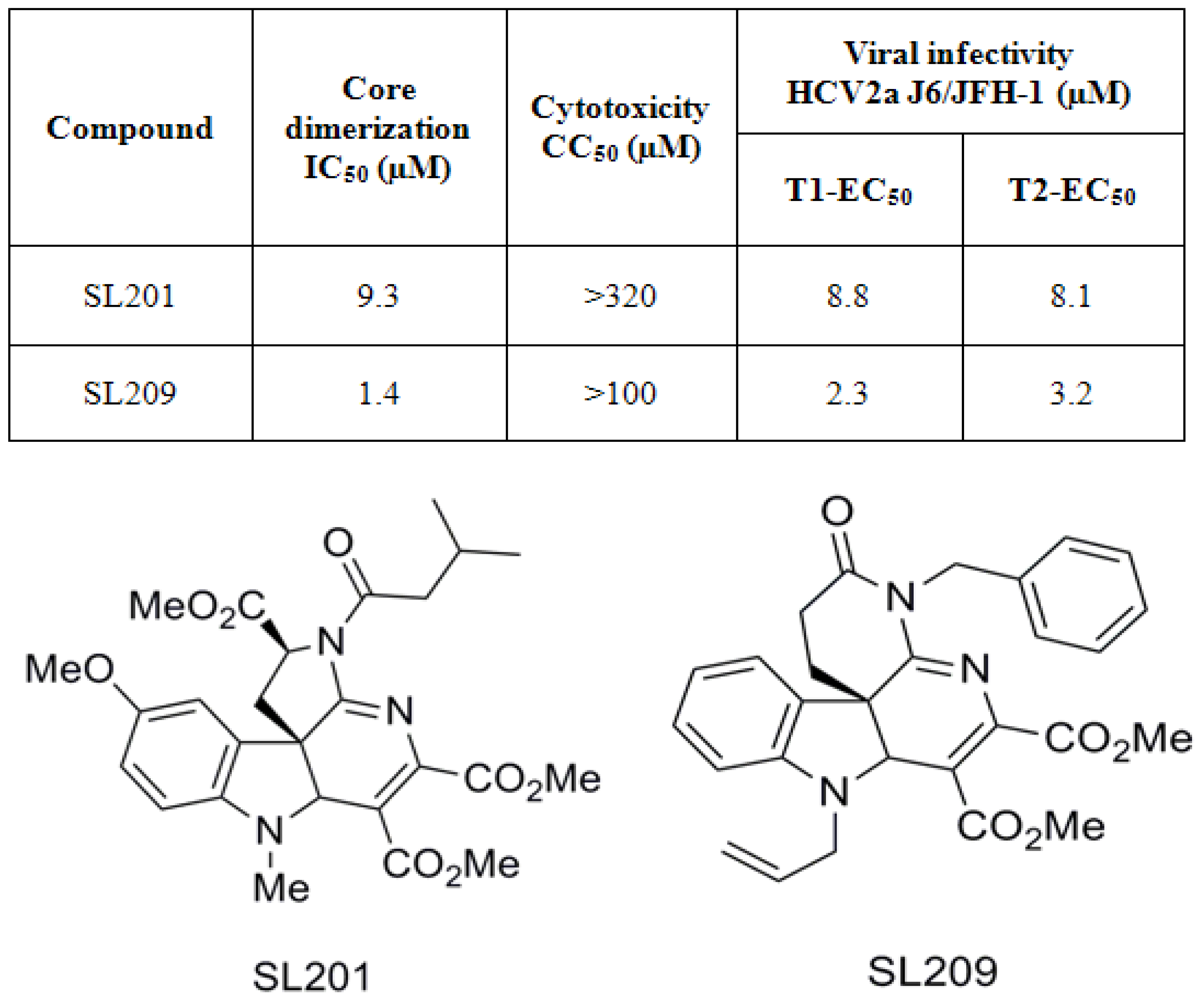
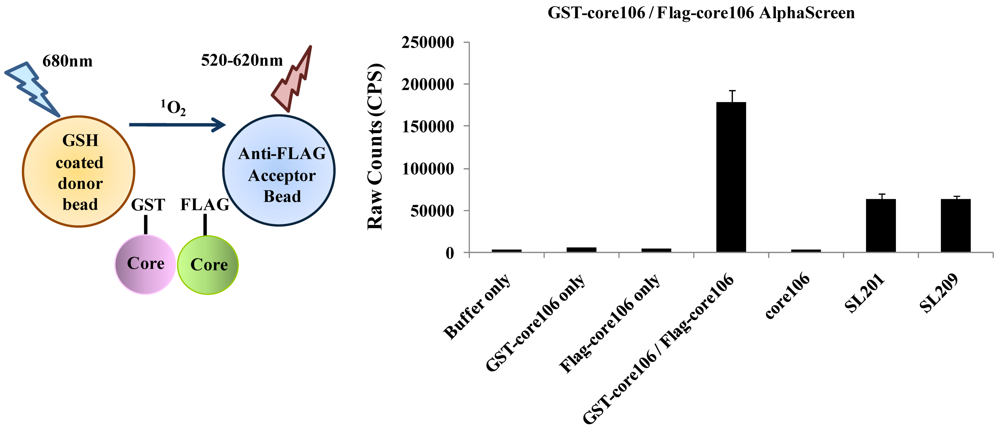
- 3. To develop a more sensitive assay for precise dose-response analyses, an AlphaScreen assay was developed, as a secondary format for monitoring core106 dimerization, but using donor and acceptor beads coated respectively with Glutathione or anti-Flag antibodies respectively, resulting in assays with at least 50-fold signal to background ratios (Figure 5) [23,70]. Again, untagged core106 domain was used as a reference inhibitor, with an IC50 of 89 nM [23]. While AlphaScreen beads are not easily handled by most robotic suites found in high-throughput screening laboratories, and sometimes vary in day-to-day stability, they provided a remarkably sensitive format for following chemical optimization of primary hit compounds first identified in large scale screening [70].
6. Inhibitors of core dimerization
6.1. Inhibition of core106 and core169 dimerization by peptides
6.2. Inhibitors of core dimerization by organic molecules
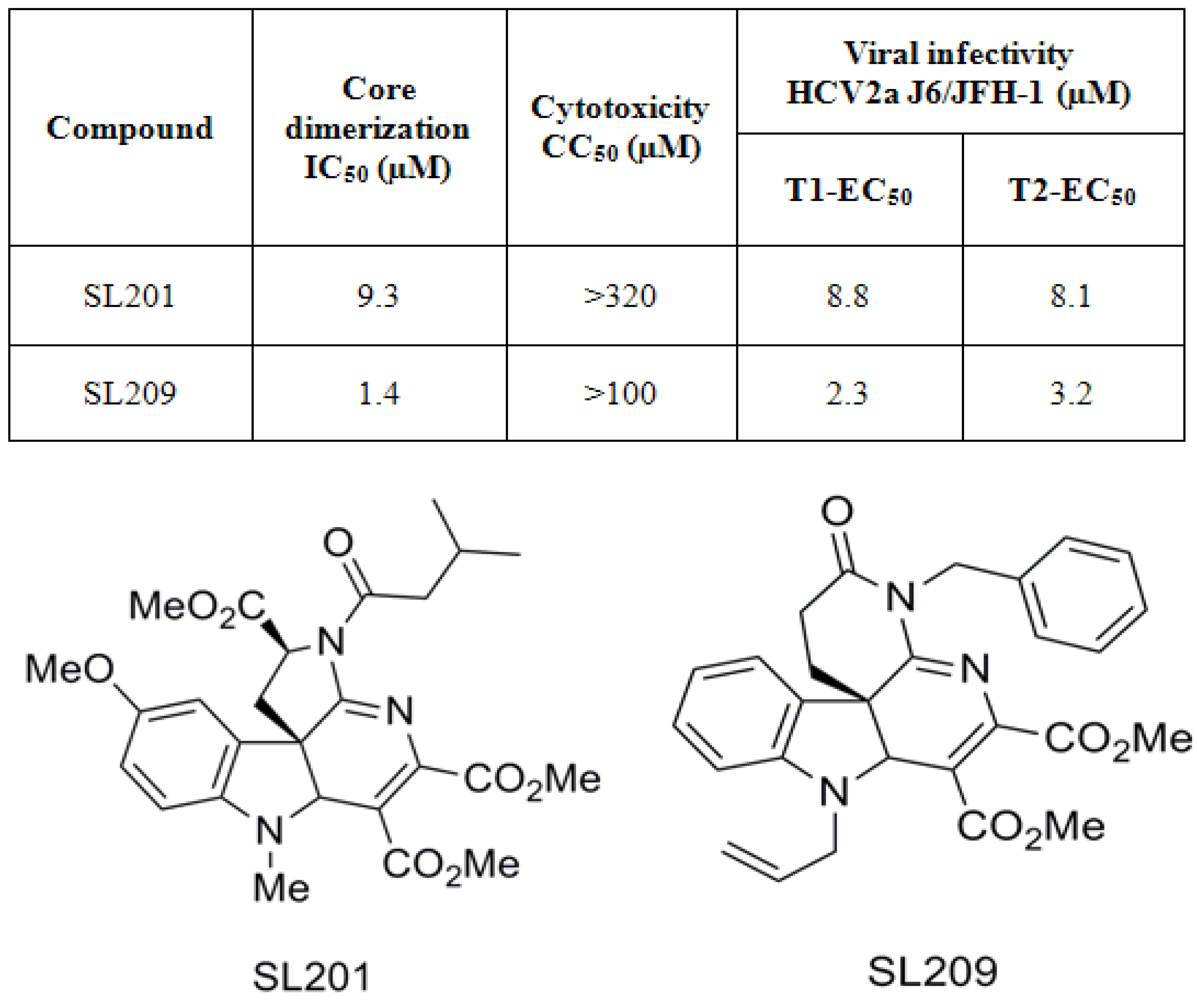
7. Inhibition of HCV production by inhibitors of core dimerization
7.1. Peptide inhibitors of HCV
7.2. Small compound inhibitors of HCV
7.3. Mode of action of inhibitors
7.3.1. Peptides
7.3.2. Small organic molecule inhibitors
8. Future developments
Acknowledgments
References
- Alter, M.J. Epidemiology of Hepatitis C infection World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar]
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.; Myers, G.; Howard, C.; Brettin, T.; Bukh, J.; Gaschen, B.; Gojobori, T.; Maertens, G.; Mizokami, M.; Nainan, O.; Netesov, S.; Nishioka, K.; Shini, T.; Simmonds, P.; Smith, D.; Stuyver, L.; Weiner, A. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch. Virol. 1998, 143, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Holmes, E.C.; Cha, T.A.; Chan, S.W.; McOmish, F.; Irvine, B.; Beall, E.; Yap, P.L.; Kolberg, J.; Urdea, M.S. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J. Gen. Virol. 1993, 74, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: The viruses and their replication. In Fields Virology, 45th; Volume 1, Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2007; pp. 1101–1152. [Google Scholar]
- Giannini, C.; Brechot, C. Hepatitis C virus biology . Cell Death Differ. 2003, 10, S27–S38. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.; Kuo, G.; Weiner, A.; Overby, L.; Bradley, D.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome . Science 1989, 244, 359–365. [Google Scholar] [PubMed]
- Dubuisson, J. Hepatitis C virus proteins. World J. Gastroenterol. 2007, 13, 2406–2415. [Google Scholar] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Cristina, J.; Moreno-del Pilar, M.; Moratorio, G. Hepatitis C virus genetic variability in patients undergoing antiviral therapy. Virus Res. 2007, 127, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Meurs, E.F.; Breiman, A. The interferon inducing pathways and the hepatitis C virus. World J. Gastroenterol. 2007, 13, 2446–2454. [Google Scholar] [PubMed]
- Wang, P.; Chun, B.K.; Rachakonda, S.; Du, J.; Khan, N.; Shi, J.; Stec, W.; Cleary, D.; Ross, B.S.; Sofia, M.J. An efficient and diastereoselective synthesis of PSI-6130: a clinically efficacious inhibitor of HCV NS5B polymerase. J. Org. Chem. 2009, 74, 6819–6824. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.; Asmuth, D.; Vargas, H.; DubucPatrick, G.; Liu, W.; Pietropaolo, K.; Zhou, X.J.; Suulivan-Bolyal, J.; Mayers, D.; The IDX -08C-003 investigator group. Antiviral Activity, Pharmacokinetics and Safety of IDX184 in Combination with Pegylated Interferon (pegIFN) and Ribavirin (RBV) in Treatment-Naive HCV Genotype 1-infected Subjects . EasL 2010, 4, 15.10. [Google Scholar]
- Ruebsam, F.; Tran, C.V.; Li, L.S.; Kim, S.H.; Xiang, A.X.; Zhou, Y.; Blazel, J.K.; Sun, Z.; Dragovich, P.S.; Zhao, J.; McGuire, H.; Murphy, D.E.; Tran, M.T.; Stankovic, N.; Ellis, D.A.; Gobbi, A.; Showalter, R.E.; Webber, S.E.; Shah, A.M.; Tsan, M.; Patel, R.A.; LeBrun, L.A.; Hou, H.J.; Kamran, R.; Sergeeva, M.V.; Bartkowski, D.M.; Nolan, T.G.; Norris, D.A.; Kirkovsky, L. 5,6-Dihydro-1H-pyridin-2-ones as potent inhibitors of HCV NS5B polymerase . Bioorg. Med. Chem. Lett. 2009, 19, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Carleo, M.A.; Borgia, F.; Castaldo, G.; Borgia, G. The efficacy and safety of telaprevir - a new protease inhibitor against hepatitis C virus. Expert Opin. Investig. Drugs 2010, 19, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Prevelige Jr., P.E. Inhibiting Virus Capsid Assembly by Altering the Polymerization Pathway. Trends Biotechnol. 1998, 16, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Bourne, C.R.; Finn, M.G.; Zlotnick, A. Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. J. Virol. 2006, 80, 11055–11061. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Sticht, J.; Humbert, M.; Findlow, S.; Bodem, J.; Müller, B.; Dietrich, U.; Werner, J.; Kräusslich, H.G. A peptide inhibitor of HIV-1 assembly in-vitro. Nat. Struct. Mol. Biol. 2005, 12, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Akuta, N.; Suzuki, F.; Hirakawa, M.; Kawamura, Y.; Yatsuji, H.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Arase, Y.; Ikeda, K.; Chayama, K.; Nakamura, Y.; Kumada, H. Amino acid substitution in HCV core region and gentic variation near IL28B gene predict viral reposne to telaprevir with peginterferon and ribavirin . Hepatology 2010, Preprint mar 2010. [Google Scholar]
- Courcambeck, J.; Bouzidi, M.; Perbost, R.; Jouirou, B.; Amrani, N.; Cacoub, P.; Pepe, G.; Sabatier, J.M.; Halfon, P. Resistance of hepatitis C virus to NS3-4A protease inhibitors: mechanisms of drug resistance induced by R155Q, A156T, D168A and D168V mutations. Antivir. Ther. 2006, 11, 847–855. [Google Scholar] [PubMed]
- De Francesco, R.; Carfi, A. Advances in the development of new therapeutic agents targeting the NS3-4A serine protease or the NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Adv. Drug Delivery. Rev. 2007, 59, 1242–1262. [Google Scholar] [CrossRef]
- Kota, S.; Coito, C.; Mousseau, G.; Lavergne, J-P; Strosberg, A.D. Peptide inhibitors of Hepatitis C Virus core oligomerization and virus production . J. Gen. Virol. 2009, 90, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.Y.; Selby, M.J.; Ou, J.H. Interaction between hepatitis C virus core protein and E1 envelope protein. J. Virol. 1996, 70, 5177–5182. [Google Scholar] [PubMed]
- Murray, C.L.; Jones, C.T.; Tassello, J.; Rice, C.M. Alanine Scanning of the Hepatitis C Virus Core Protein Reveals Numerous Residues Essential for Infectious Virus Production. J. Virol. 2007, 81, 10220–10231. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J. Lipid droplets and hepatitis C infection. Biochim. Biophys. Acta 2009, 1791, 552–559. [Google Scholar] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production . Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Flajolet, M.; Rotondo, G.; Daviet, L.; Bergametti, F.; Inchauspe, G.; Tiollais, P.; Transy, C.; Legrain, P. A genomic approach of the hepatitis C virus generates a protein interaction map. Gene 2000, 242, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Goh, P.Y.; Tan, Y.J.; Lim, S.P.; Lim, S.G.; Tan, Y.H.; Hong, W.J. The hepatitis C virus core protein interacts with NS5A and activates its caspase-mediated proteolytic cleavage. Virology 2001, 290, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, M.; Imbert, I.; Kieny, M.P. Schuster,C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 2003, 77, 5401–5414. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; Suzuki, T. Interaction of Hepatitis C Virus Nonstructural Protein 5A with Core Protein Is Critical for the Production of Infectious Virus Particles J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, 1000035. [Google Scholar] [CrossRef]
- Duvignaud, J.B.; Savard, C.; Fromentin, R.; Majeau, N.; Leclerc, D.; Gagné, S.M. Structure and dynamics of the N-terminal half of hepatitis C virus core protein: An intrinsically unstructured protein. Biochem. Biophys. Res. Commun. 2008, 378, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Barba, G.; Harper, F.; Harada, T.; Kohara, M.; Goulinet, S.; Matsuura, Y.; Eder, G.; Schaff, Z.; Chapman, M.J.; Miyamura, T.; Brechot, C. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Shavinskay, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly . J.Biol.Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Santolini, E.; Migliaccio, G.; LaMonica, N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 1994, 68, 3631–3641. [Google Scholar] [PubMed]
- Hüssy, P.; Langen, H.; Mous, J.; Jacobsen, H. Hepatitis C Virus Core Protein: Carboxy-Terminal Boundaries of Two Processed Species Suggest Cleavage by a Signal Peptide Peptidase. Virology 1996, 224, 93–104. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Lee, Y.W.; Chen, S.S. Characterization of hepatitis C virus core protein multimerization and membrane envelopment: revelation of a cascade of core-membrane interactions. J. Virol. 2009, 83, 9923–9939. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 2007, 81, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.I.; Dubuisson, J. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol. Cell 2009, 102, 63–74. [Google Scholar]
- Targett-Adams, P.; Boulant, S.; Douglas, M.W.; McLauchlan, J. Lipid Metabolism and HCV Infection. Viruses 2010, 2, 1195–1217. [Google Scholar] [CrossRef]
- Kopp, M.; Murray, C.L.; Jones, C.T.; Rice, C.M. Genetic analysis of the carboxy –terminal region of the Hepatitis C virus core protein. J. Virol. 2010, 84, 1666–1673. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J. Hepatitis C virus: viral proteins on the move. Biochem. Soc. Trans. 2009, 37, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Depla, M.; Uzbekov, R.; Hourioux, C.; Blanchard, E.; LeGouge, A.; Gillet, L.; Roingeard, P. Ultrastructural and quantitative analysis of the lipid droplet clustering induced by hepatitis C virus core protein . Cell Mol. Life Sci. 2010. [Google Scholar] [PubMed]
- Kim, M.; Ha, Y.; Park, H.J. Structural requirements for assembly and homotypic interactions of the hepatitis C virus core protein. Virus Res. 2006, 122, 137–43. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Targett-Adams, P.; McLauchlan, J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 2007, 88, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Cristofari, G.; Ivanyi-Nagy, R.; Gabus, C.; Boulant, S.; Lavergne, J.P.; Penin, F.; Darlix, J.L. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004, 32, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Ivanyi-Nagy, R.; Kanevsky, I.; Gabus, C.; Lavergne, J.P.; Ficheux, D.; Penin, F.; Fosse, P.; Darlix, J.L. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006, 34, 2618–2633. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Montserret, R.; Hope, R.G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.P.; Penin, F.; McLauchlan, J. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Vanabelle, C.; Ebel, C.; Penin, F.; Lavergne, J.P. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J. Virol. 2005, 79, 11353–11365. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, M.K.; Martoglio, B. Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol. Cell. 2002, 10, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Mori, Y.; Komoda, Y.; Okamoto, T.; Okochi, M.; Takeda, M.; Suzuki, T.; Morishi, K.; Matsuura, Y. Intramembrane processing by signal peptide peptidase regulates the membrane localization of hepatitis C virus core protein and viral propagation. J. Virol. 2008, 82, 8349–8361. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Wakita, T.; Tsukiyama-Kohara, K.; Funahashi, S.-I.; Ichikawa, M.; Kajita, T.; Moradpour, D.; Wands, J.R.; Kohara, M. The Native Form and Maturation Process of Hepatitis C Virus Core Protein. J. Virol. 1998, 72, 6048–6055. [Google Scholar] [PubMed]
- Kunkel, M.; Lorinczi, M.; Rijnbrand, R.; Lemon, S.M.; Watowich, S.J. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J. Virol. 2001, 75, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Targett-Adams, P.; McLauchlan, J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 2007, 88, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Majeau, N.; Laliberte Gagne, M.E.; Boivin, A.; Duvignaud, J.B.; Leclerc, D.A. method for in vitro assembly of hepatitis C virus core protein and for screening of inhibitors. Anal. Biochem. 2007, 366, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hsieh, T.Y.; Zhu, N.; VanArsdale, T.; Hwang, S.B.; Jeng, K.S.; Gorbalenya, A.E.; Lo, S.Y.; Ou, J.H.; Ware, C.F.; Lai, M.M. Hepatitis C virus core protein interacts with the cytoplasmic tail of lymphotoxin-beta receptor. J. Virol. 1997, 71, 1301–1309. [Google Scholar] [PubMed]
- Nolandt, O.; Kern, V.; Muller, H.; Pfaff, E.; Theilmann, L.; Welke, R.; Krausslich, H.G. Analysis of hepatitis C virus core protein interaction domains. J. Gen. Virol. 1997, 78, 1331–1340. [Google Scholar] [PubMed]
- Klein, K.C.; Polyak, S.J.; Lingappa, J.R. Unique features of hepatitis C virus capsid formation revealed by de novo cell-free assembly. J. Virol. 2004, 78, 9257–9269. [Google Scholar] [CrossRef] [PubMed]
- Majeau, N.; Gagne, V.; Boivin, A.; Bolduc, M.; Majeau, J.A.; Ouellet, D.; Leclerc, D. The N-terminal half of the core protein of hepatitis C virus is sufficient for nucleocapsid formation. J. Gen. Virol. 2004, 85, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Majeau, N.; Fromentin, R.; Savard, C.; Duval, M.; Tremblay, M.J.; Leclerc, D. Palmitoylation of hepatitis C virus core protein is important for virion production. J. Biol. Chem. 2009, 85, 971–981. [Google Scholar]
- Moriya, K.; Fujie, H. The Core Protein of Hepatitis C Virus Induces Hepatocellular Carcinoma in Transgenic Mice. Nat. Med. 1998, 4, 1065–1069. [Google Scholar] [CrossRef]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78, 1527–1531. [Google Scholar] [PubMed]
- Kota, S.; Scampavia, L.; Spicer, T.; Beeler, A.B.; Takahashi, V.; Snyder, J.K.; Porco, J.A.Jr.; Hodder, P.; Strosberg, A.D. A homogeneous time resolved fluorescence assay for identifying inhibitors of Hepatitis C virus dimerization. Assay Drug Dev. Technol. 2010, 8, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Cai, C.; Kota, S.; Takahashi, V.; Strosberg, A.D.; Snyder, J.K. New Small Molecule Inhibitors of Hepatitis C Virus Bioorg. Med. Chem. Lett. 2009, 19, 6926–6930. [Google Scholar] [CrossRef] [Green Version]
- Peppard, J.; Glickman, F.; He, Y.; Hu, S.I.; Doughty, J.; Goldberg, R. Development of a high-throughput screening assay for inhibitors of aggrecan cleavage using luminescent oxygen channeling (AlphaScreen). J. Biomol. Screen. 2003, 8, 149–156. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Nolandt, O.; Kern, V.; Muller, H.; Pfaff, E.; Theilmann, L.; Welker, R.; Krausslich, H.G. Analysis of hepatitis C virus core protein interaction domains . J. Gen. Virol. 1997, 78, 1331–1340. [Google Scholar] [PubMed]
- Mousseau, G.; Strosberg, A.D. Personal communication . 2008. [Google Scholar]
- Cordell, G.A. The Aspidosperma Alkaloids. The Alkaloids 1978, 17, 199–384. [Google Scholar]
- Saxton, J.E. Alkaloids of the Aspidospermine Group. In The Alkaloids; Cordell, G.A., Ed.; Academic Press: New York, NY, USA, 1998. [Google Scholar]
- Benson, S.C.; Lee, L.; Yang, L.; Snyder, J.K. Intramolecular Inverse Electron Demand Diels-Alder Reactions of Tryptamine with Tethered Heteroaromatic Azadienes. Tetrahedron 2000, 56, 1165–1180. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Montero, A.; Gastaminza, P.; Whitten-Bauer, C.; Wieland, S.F.; Isogawa, M.; Fredericksen, B.; Selvarajah, S.; Gallay, P.A.; Ghadiri, M.R.; Chisari, F.V. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3088–3093. [Google Scholar] [CrossRef] [PubMed]
- Heller, T.; Saito, S.; Auerbach, J.; Williams, T.; Moreen, T.R.; Jazwinski, A.; Cruz, B.; Jeurkar, N.; Sapp, R.; Luo, G.; Liang, T.J. An in vitro model of hepatitis C virion production. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 2579–2583. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Strosberg, A.D.; Kota, S.; Takahashi, V.; Snyder, J.K.; Mousseau, G. Core as a Novel Viral Target for Hepatitis C Drugs. Viruses 2010, 2, 1734-1751. https://doi.org/10.3390/v2081734
Strosberg AD, Kota S, Takahashi V, Snyder JK, Mousseau G. Core as a Novel Viral Target for Hepatitis C Drugs. Viruses. 2010; 2(8):1734-1751. https://doi.org/10.3390/v2081734
Chicago/Turabian StyleStrosberg, Arthur Donny, Smitha Kota, Virginia Takahashi, John K. Snyder, and Guillaume Mousseau. 2010. "Core as a Novel Viral Target for Hepatitis C Drugs" Viruses 2, no. 8: 1734-1751. https://doi.org/10.3390/v2081734
APA StyleStrosberg, A. D., Kota, S., Takahashi, V., Snyder, J. K., & Mousseau, G. (2010). Core as a Novel Viral Target for Hepatitis C Drugs. Viruses, 2(8), 1734-1751. https://doi.org/10.3390/v2081734