The IKK Kinases: Operators of Antiviral Signaling

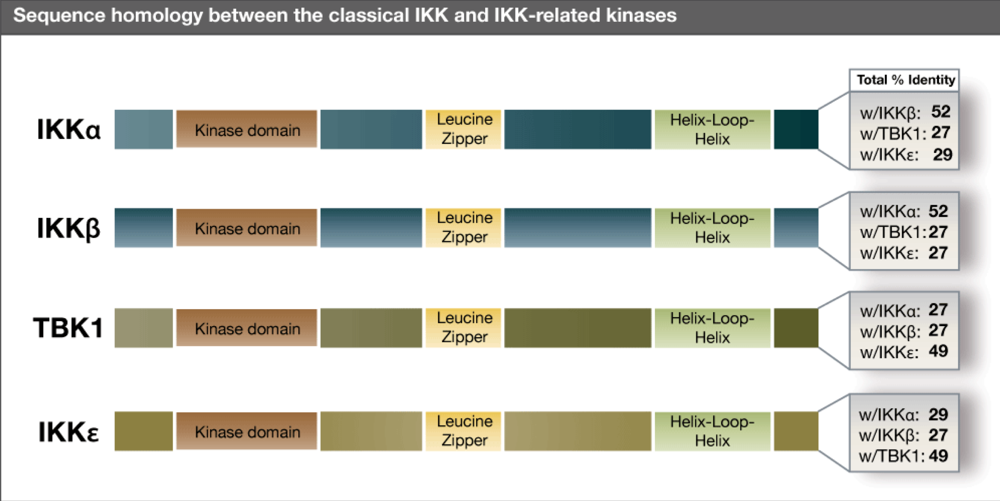
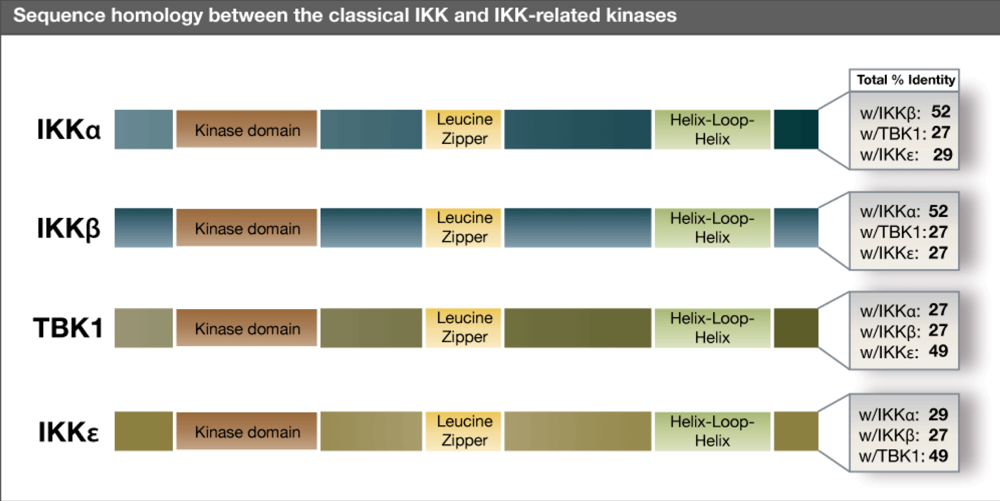{kind=link}
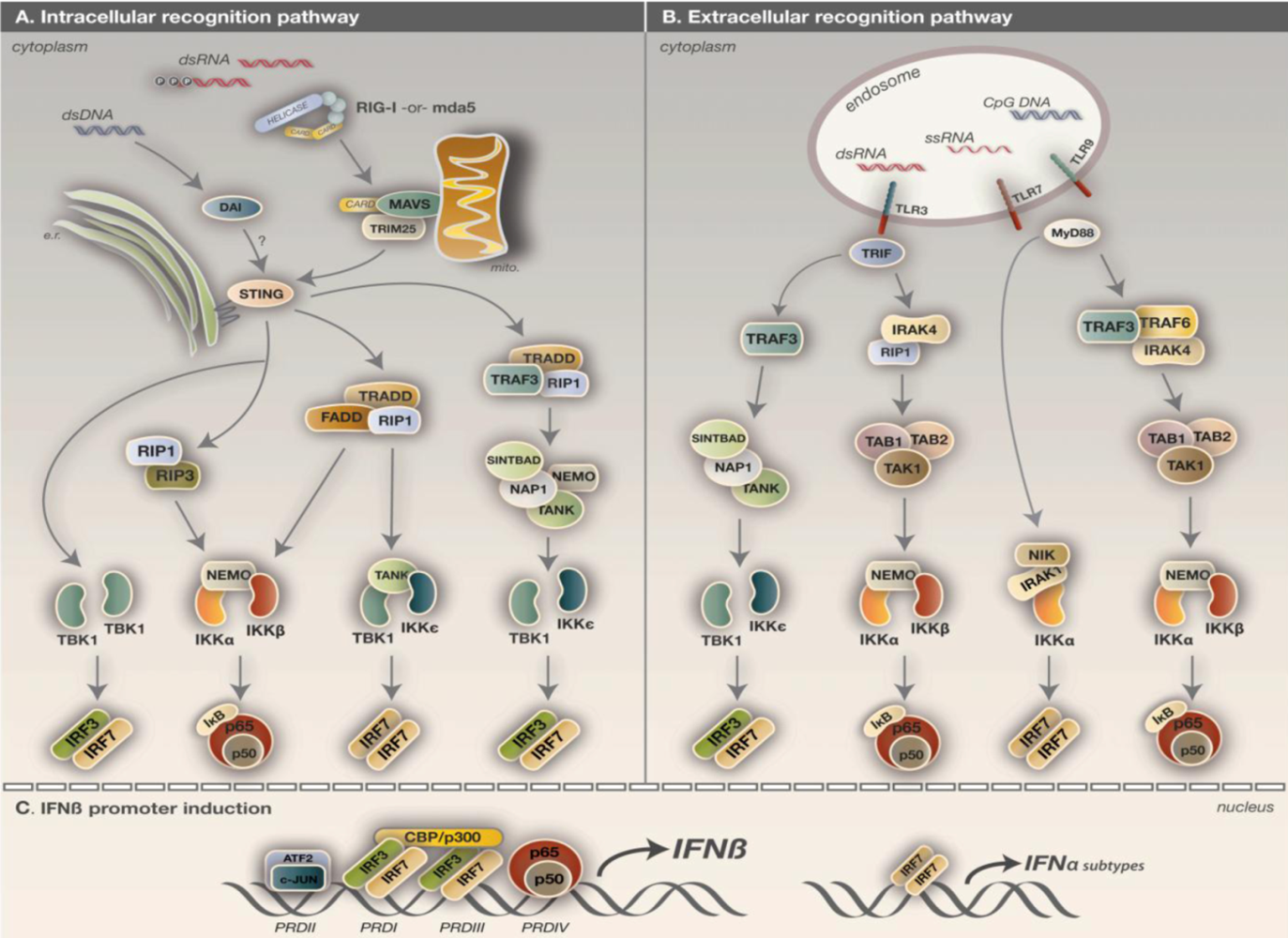{kind=link}
Abstract
:1. Introduction
2. Transcriptional regulation of the IFNβ promoter
3. The IKK complex
5. Detection of virus infection
5.1. IKK activation via the TLR pathway
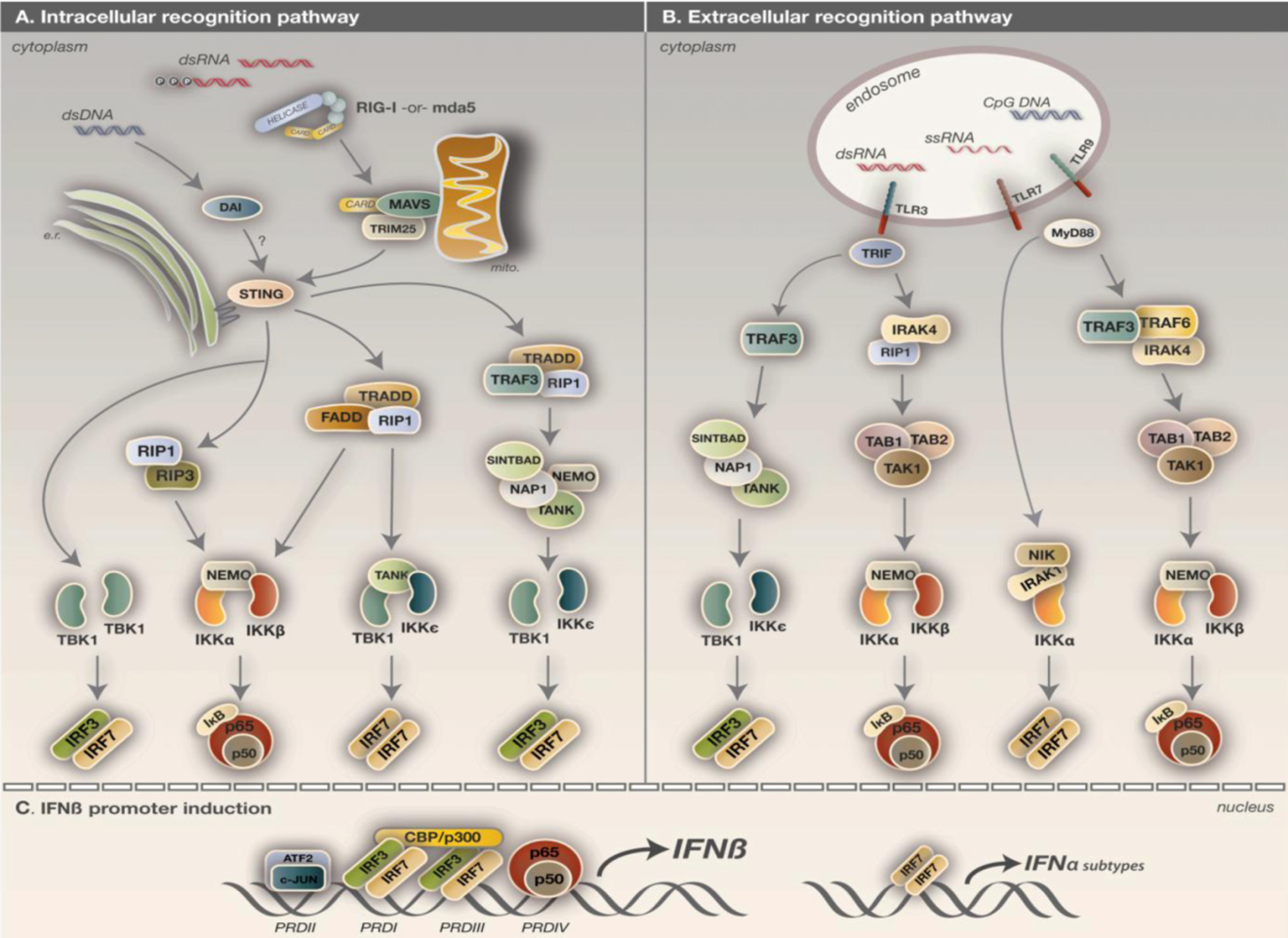
5.2. IKK activation via the RLH pathway
5.3. IKK activation via the DNA sensory pathway
5.4. IKK activation via IFN signaling
7. Conclusion
References
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, T.; Falvo, J.V.; Kim, T.H.; Kim, T.K.; Lin, C.H.; Parekh, B.S.; Wathelet, M.G. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 609–620. [Google Scholar] [PubMed]
- Stark, G.; Kerr I.; Williams B.; Silverman, R.; Schreiber R. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Stark, G. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.; tenOever, B.; Maniatis, T. Connecting mitochondria and innate immunity. Cell 2005, 122, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 357, re13. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.; Grandvaux, N.; Zhou, G.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.; McWhirter, M.; Faia,K.; Rowe, D.; Latz, E.; Golenbock, D.; Coyle, A.; Liao, S.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Panne, D.; Maniatis, T.; Harrison, S. An atomic model of the interferon-beta enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Structure of IRF-3 bound to the PRDIII-I regulatory element of the human interferon-beta enhancer. Mol. Cell 2007, 26, 703–716. [CrossRef] [PubMed]
- Panne, D.; Maniatis, T.; Harrison, S. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-beta enhancer. EMBO J. 2004, 23, 4384–4393. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- NF-kappa B: a lesson in family values. Cell 1995, 80, 529–532. [CrossRef] [PubMed]
- Chen, Z.; Parent, L.; Maniatis, T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell 1996, 84, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Fujita, T.; Miyamoto, M.; Furia, A.; Miyata, T.; Taniguchi, T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 1989, 58, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Mamane, Y.; Heylbroeck, C.; Genin, P.; Algarte, M.; Servant, M.; LePage, C.; DeLuca, C.; Kwon, H.; Lin, R.; Hiscott, J. Interferon regulatory factors: the next generation. Gene 1999, 237, 1–14. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.Ogasawara; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sealfon, S.; Hayot, F.; Jayaprakash, C.; Kumar, M.; Pendelton, A.; Ganee, A.; Fernandez-Sesma, A.; Moran, T. Chromosome-specific and noisy IFNB1 transcription in individual virus-infected human primary dendritic cells. Nucleic Acids Res. 2007, 35, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Genin, P.; Mamane, Y.; Hiscott, J. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 2000, 20, 6342–6353. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Mamane, Y.; Hiscott, J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 2000, 275, 34320–34327. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Heylbroeck, C.; Pitha, P.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [PubMed]
- Servant, M.; Grandvaux, N.; tenOever, B.; Duguay, D.; Lin, R.; Hiscott, J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 2003, 278, 9441–9447. [Google Scholar] [CrossRef] [PubMed]
- Clement, J; Bibeau-Poirier, A.; Gravel, S.; Grandvaux, N.; Bonneli, E.; Thibault, P.; Meloche, S.; Servant, M. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. J. Virol. 2008, 82, 3984–3996. [Google Scholar] [CrossRef] [PubMed]
- Zandi, E.; Rothwarf, D.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka S, G Courtois; Weil, R.; Agou, F.; Kirk, H.; Kay, R.; Israel, A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 1998, 93, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Rothwarf, D.; Zandi, E.; Natoli, G.; Karin, M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.; Shevchenko, A.; Bennet, B.; Li, J.; Young, D.; Barbosa, M.; Mann, M.; Manning, A.; Rao, A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Solt, LA.; Madge, LA.; May, MJ. NEMO-binding domains of both IKKalpha and IKKbeta regulate IkappaB kinase complex assembly and classical NF-kappaB activation. J. Biol. Chem. 2009, 284, 27596–27608. [Google Scholar] [CrossRef] [PubMed]
- Chau TL, R Gioia; Carpentier, I.; Chapelle, J.; O'Neill, L.; Beyaert, R.; Piette, J.; Chariot, A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem. Sci. 2008, 33, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Leun, T.; Hoffmann, A.; Baltimore, D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell 2004, 118, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Ostertag, D.; Li, Z.; Chang, L.; Chen, Y.; Hu, Y.; Williams, B.; Perrault, J.; Karin, M. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity 1999, 11, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, FR.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, SC.; Karin, M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappaB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Bebien, M.; Otero, D.; Johnson-Vroom, K.; Cao, Y.; Vu, D.; Jegga, A.; Aronow, B.; Ghosh, G.; Rickert, R.; Karin, M. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Sugiyama, T.; Matsumoto, M.; Tanaka, T.; Saito, M.; Hemmi, H.; Ohara, O.; Akira, S.; Kaisho, T. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 2006, 440, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Shultz, D.; Fuller, J.; Yang, Y.; Sizemore, N.; Rani, M.; Stark, G. Activation of a subset of genes by IFN-gamma requires IKKbeta but not interferon-dependent activation of NF-kappaB. J. Interferon Cytokine Res. 2007, 27, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, X,; Hussain, S.; Wang, E.; Wang, X.; Li, M.; Garcia-Sastre, A.; Beg A. Lack of essential role of NF-kappa B p50, RelA, and cRel subunits in virus-induced type 1 IFN expression. J. Immunol. 2007, 178, 6770–6776. [Google Scholar] [PubMed]
- Massa, P.; Li, X.; Hanidu, A.; Siamas, J.; Pariali, M.; Pareja, J.; Savitt, A.; Catron, J.; Li, J.; Marcu, K. Gene expression profiling in conjunction with physiological rescues of IKKalpha-null cells with wild type or mutant IKKalpha reveals distinct classes of IKKalpha/NF-kappaB-dependent genes. J. Biol. Chem. 2005, 280, 14057–14069. [Google Scholar] [CrossRef] [PubMed]
- Bonnard M, C Mirtsos; Huang, J.; Ng, M.; Itie, A.; Wakeham, A.; Shahinian, A.; Henzel, W.; Elia, A.; Shillinglaw, W.; Mak, T.; Cao, Z. Yeh. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000, 19, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Shimada T, T Kawai; Inoue, J.; Tatsumi, Y.; Kanamaru, A.; Akira, S. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 1999, 11, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.; Maniatis, T. A new family of IKK-related kinases may function as I kappa B kinase kinases. Biochim. Biophys. Acta. 2001, 1471, M57–M62. [Google Scholar] [PubMed]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Chow, E.; Goodnough, J.; Yeh, W.; Cheng, G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 2004, 199, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Panne, D.; McWhirter, S.; Maniatis, T.; Harrison, S. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 2007, 282, 22816–22822. [Google Scholar] [CrossRef] [PubMed]
- tenOever, B.; Ng, S.; Chua, M.; McWhirter, S.; Garcia-Sastre, A; Maniatis, T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- tenOever, B.; Sharma, S.; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.; Lin, R.; Hiscott, J. Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 2004, 78, 10636–10649. [Google Scholar] [CrossRef] [PubMed]
- Hutti, J.; Shen, R.; Abbott, D.; Zhou, A.; Sprott, K.; Asara, J.; Hahn, W.; Cantley, L. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol. Cell 2009, 34, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.; O'Donnell, M.; Legarda-Addison, D.; Ng, A.; Cardenas, W.; Yount, J.; Moran, T.; Basler, C.; Komuro, A.; Horvath, C.; Xavier, R.; Ting, A. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Kravchenk, V.; Mathison, J.; Schwamborn, K.; Mercurio, F.; Ulevitch, R. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J. Biol. Chem. 2003, 278, 26612–26619. [Google Scholar] [CrossRef] [PubMed]
- Adli, M.; Baldwin, A. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 2003, 281, 26976–26984. [Google Scholar] [CrossRef]
- Harris, J.; Oliere, S.; Sharma, S.; Sun, Q.; Lin, R.; Hiscott, J.; Grandvaux, N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J. Immunol. 2006, 177, 2527–2535. [Google Scholar] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, T.; Sato, S.; Matsushita, K.; Kato, H.; Matsui, K.; Kumagai, Y.; Saitoh, T.; Kawai, T.; Takeuchi, O.; Akira, S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 2008, 9, 684–691. [Google Scholar] [CrossRef]
- Suzuki, N.; Suzuki, S.; Duncan, G.; Miller, D.; Wada, T.; Mirtsos, C.; Takada, H.; Wakeham, A.; Itie, A.; Li, S.; Penninger, J.; Wesche, H.; Ohashi, P.; Mak, T.; Yeh, W. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 2002, 416, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Sato, S.; Yamamoto, M.; Hirotani, T.; Kato, H.; Takeshita, F; Matsuda, M.; Coban, C.; Ishii, K.; Kawai, T.; Takeuchi, O.; Akira, S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 2005, 201, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, M.; Li, Y.; Diao, F.; Chen, D.; Zhai, Z.; Shu, H. Differential regulation of IKK alpha-mediated activation of IRF3/7 by NIK. Mol. Immunol. 2008, 45, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Cusson-Hermance, N.; Khurana, S.; Lee, T.; Fitzgerald, K.; Kelliher, M. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J. Biol. Chem. 2005, 280, 36560–36566. [Google Scholar] [CrossRef] [PubMed]
- Gohda, J.; Matsumura, T.; Inoue, J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J. Immunol. 2004, 173, 2913–2917. [Google Scholar] [PubMed]
- Baud, V.; Liu, Z.; Bennett, B.; Suzuki, N.; Xia, Y.; Karin, M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999, 13, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, SK.; Guo, B.; He, JQ.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Ryzhakov, G.; Randow, F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007, 26, 3180–3190. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Locarnini, S. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell Biol. 2007, 85, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, C.; Stewart, M.; Xu, H.; Strong, R.; Igumenova, T.; Li, P. Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch. Biochem. Biophys. 2009, 488, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G; Juranek, S.; Kato, H.; Kawai, T.; Poeck, H.; Fitzgerald, K.; Takeuchi, O.; Akira, S.; Tuschl, T.; Latz, E.; Ludwig, J.; Hatmann, G. Recognition of 5' triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Sun, L.; Ea, C.; Chen, Z. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D Binder; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.; Han, K.; Ly, L.; Zhai, Z.; Shu, H. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.; Shin, Y.; Joo, C.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; Jung, J. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Pietras, E.; He, J.; Kang, J.; Liu, S.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.; Wang, Y.; Cheng, G. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Thomas, E.; Barber, G. A FADD-dependent innate immune mechanism in mammalian cells. Nature 2004, 432, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Michallet, M.; Meylan, E.; Ermolaeva, M.; Vasquez, J.; Rebasamen, M.; Curran, J.; Poeck, H.; Bscheider, M.; Hartmann, G.; Konig, M.; Kalinke, U.; Pasparakis, M.; Tschopp, J. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 2008, 28, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J. Immunol. 2006, 176, 4520–4524. [Google Scholar] [PubMed]
- Balachandran, S.; Venkataraman, T.; Fisher, P.; Barber, G. Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7. J. Immunol. 2007, 178, 2429–2439. [Google Scholar] [PubMed]
- Arnoult, D.; Carneiro, L.; Tattoli, I.; Girardin, S. The role of mitochondria in cellular defense against microbial infection. Semin. Immunol. 2009, 21, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Paz S, M Vilasco; Lacoste, J.; Nguyen, T.; Zhao, T.; Shestakova, E.; Zaari, S.; Bibeau-Poirier, A.; Servant, M.; Lin, R.; Meurs, E.; Hiscott, J. Ubiquitin-regulated recruitment of IkappaB kinase epsilon to the MAVS interferon signaling adapter. Mol. Cell. Biol. 2009, 29, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yang, L.; Sun, Q.; Arguello, M.; Ballard, D.; Hiscott, J.; Lin, R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 2007, 8, 592–600. [Google Scholar] [CrossRef]
- Moore, C.; Bergstralh, D.; Duncan, J.; Lei, Y.; Morrison, T.; Zimmerman, A.; Accavitti-Loper, M.; Madden, V.; Sun, L.; Ye, Z.; Lich, J.; Heise, M.; Chen, Z.; Ting, J. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Heinz, L.; Meylan, E.; Michallet, M.; Schroder, K.; Hofmann, K.; Vasquez, J.; Benedict, C.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 8, 916–922. [Google Scholar] [CrossRef]
- Roberts, T.; Idris, A.; Dunn, J.; Kelly, G.; Burnton, C.; Hodgson, S.; Hardy, L.; Garceau, V.; Sweet, M.; Ross, I.; Hume, D.; Stacey, K. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, C.; Caffrey, D.; Latz, E.; Fitzgerald, K. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.; Datta, P.; Wu, J.; Alnemri, E. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.; Upton, J.; Mocarski, E. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [PubMed]
- Miyahira, A.; Shahangian, A.; Hwang, S.; Sun, R.; Cheng, G. TANK-binding kinase-1 plays an important role during in vitro and in vivo type I IFN responses to DNA virus infections. J. Immunol. 2009, 182, 2248–2257. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.; Petrilli, V.; Zaiss, A.; White, L.; Clark, S.; Ross, P.; Parks, R.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, N.; Agarwal, A.; Das, K.; Lerner, N.; Sulak, M.; Rani, S.; Ransohoff, R.; Shultz, D.; Stark, G. Inhibitor of kappaB kinase is required to activate a subset of interferon gamma-stimulated genes. Proc. Natl. Acad. Sci. USA 2004, 101, 7994–7998. [Google Scholar] [CrossRef]
- Platanias, L. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Pomerantz,J.; Baltimore, D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Gatot, J.; Gioia, R.; Chau, T.; Patrascu, F.; Warnier, M.; Close, P.; Chapelle, J.; Muraille, E.; Brown, K.; Siebenlist, U.; Piette, J.; Dejardin, E.; Chariot, A. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J. Biol. Chem. 2007, 282, 31131–31146. [Google Scholar] [CrossRef] [PubMed]
- Fujita,F.; Taniguchi, Y.; Kato, T.; Narita, Y.; Furuya, A.; Ogawa, T.; Sakurai, H.; Joh, T.; Itoh, M.; Delhase, M.; Karin, M.; Nakanishi, M. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol. 2003, 23, 7780–7793. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Pham, A.M.; TenOever, B.R. The IKK Kinases: Operators of Antiviral Signaling. Viruses 2010, 2, 55-72. https://doi.org/10.3390/v2010055
Pham AM, TenOever BR. The IKK Kinases: Operators of Antiviral Signaling. Viruses. 2010; 2(1):55-72. https://doi.org/10.3390/v2010055
Chicago/Turabian StylePham, Alissa M., and Benjamin R. TenOever. 2010. "The IKK Kinases: Operators of Antiviral Signaling" Viruses 2, no. 1: 55-72. https://doi.org/10.3390/v2010055
APA StylePham, A. M., & TenOever, B. R. (2010). The IKK Kinases: Operators of Antiviral Signaling. Viruses, 2(1), 55-72. https://doi.org/10.3390/v2010055