Evasion of the Interferon-Mediated Antiviral Response by Filoviruses


Abstract
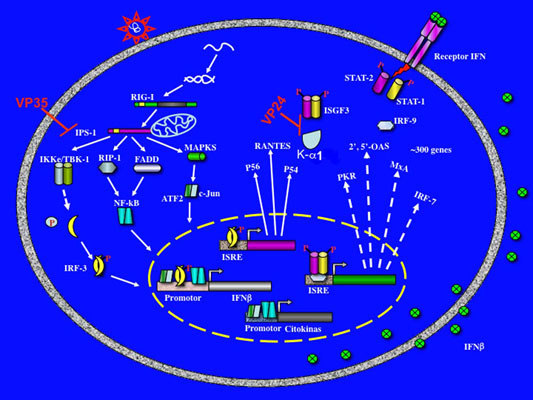
:
1. Introduction to Filovirus

{kind=link}
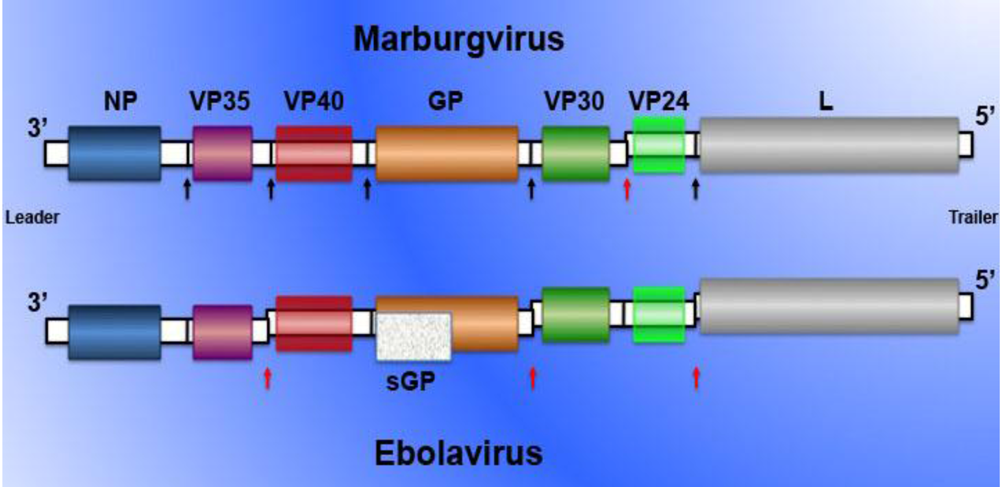{kind=link}
{kind=link}
{kind=link}
| Virus | No. | Location | Year | Human Cases (deaths) | CFR% | |
|---|---|---|---|---|---|---|
| Marburg | 1 | Germany (Marburg and Frankfurt), former Yugoslavia (Belgrade) [6]. | 1967 | 31 (7) | 23 | |
| 2 | South Africa (Johannesburg)[7]. | 1975 | 3 (1) | 33 | ||
| 3 | Kenya (Mount Elgon National Park) [6]. | 1980 | 2 (1) | 50 | ||
| 4 | Kenya (Mount Elgon National Park) [6]. | 1987 | 1 (1) | 100 | ||
| 5 | DRC (Durba, gold mine village)[8]. | 1998-2000 | 154 (128) | 83 | ||
| 6 | Angola (Uige Province) [9]. | 2004-2005 | 252 (227) | 90 | ||
| 7 | Uganda (mine workers in Kakasi Forest Reserve, Kamwenge District) [6]. | 2007 | 3 (1) | 50 | ||
| 8 | Uganda (western tourists at Maramagambo Forest)[10]. | 2008 | 2 (1) | 50 | ||
| Ebola-Zaire | 1 | DRC, formerly Zaire. (Yambuku and surroundings)[4]. | 1976 | 318 (280) | 88 | |
| 2 | DRC, formerly Zaire (Tandala Hospital, Tandala)[11]. | 1977 | 1 (1) | 100 | ||
| 3 | Gabon (Makokou General Hospital and gold-panning encampment)[12]. | 1994/1995 | 49 (29) | 59 | ||
| 4 | Gabon (outbreak began early February in the village of Mayibout 2, Gabon)[12]. | 1996 | 31 (21) | 67.7 | ||
| 5 | Gabon (outbreak started in a logging camp near Booué)[12]. | 1996/1997 | 60 (45) | 75 | ||
| 6 | DRC, formerly Zaire (outbreak centered in Kikwit and surrounding area)[13]. | 1995 | 315 (250) | 79.4 | ||
| 7 | South Africa (Imported case from Libreville, Gabon. Single fatality was the local nurse caring for the index case) WHO[14]. | 1996 | 2 (1) | 50 | ||
| 8 | Gabon and Republic of the Congo (Simultaneous outbreaks in La Zadié, Ivindo and Mpassa districts, Gabon, and Mbomo and Kéllé districts, Congo)[15]. | 2001/2002 (25 October to 18 March) | 124 (97) | 78 | ||
| 9 | Republic of the Congo (outbreak was in Mbomo district, Congo, where two fatal cases migrated to Ekata village in Gabon)[15]. | 2002 (17 May to 25 July) | 11 (10) | 91 | ||
| 10 | Republic of the Congo (outbreak was mainly present in the Kéllé district with fewer cases in the Mbomo district)[16]. | 2002/2003 (25 December to 22 April) | 143 (128) | 89.5 | ||
| 11 | Republic of the Congo (Outbreak affected the Mbomo and Mbandza villages of the Mbomo district)[17]. | 2003 | 35 (29) | 83 | ||
| 12 | Republic of the Congo (outbreak was in the west part of the country, in the Cuvette Ouest Region, towns of Etoumbi and Mbomo)[18]. | 2005 (25 April to 16 June) | 12 (9) | 75 | ||
| 13 | Democratic Republic of Congo (outbreak was in Mueka & Luebo health zones, Province of Kasai Occidental. Reports started in September 2007 until official end declaration of the outbreak on 16 February 2009)[18]. | 2007/2009 | 32 (15) | 47 | ||
| Ebola-Sudan | 1 | Sudan (Towns of Nzara, Maridi and Tembura)[5]. | 1976 | 284 (151) | 53 | |
| 2 | England (Accidental laboratory inoculation)[19]. | 1976 | 1 (0) | 0 | ||
| 3 | Sudan (Nzara and Yambio in Southern Sudan)[20] | 1979 | 34 (22) | 65 | ||
| 4 | Uganda (Outbreak initiated in the Gulu district, then spread to Mbarara and Masindi districts)[21]. | 2000/2001 | 425 (224) | 52.7 | ||
| 5 | Sudan (outbreak occurred in Yambio county, southern Sudan)[22]. | 2004 (15 April to 26 June) | 17 (7) | 41 | ||
| Ebola-Reston | 1 | USA (New EBOV in Reston, Texas, introduced with infected cynomolgus macaques from Philippines)[23]. | 1989 | 0 (0) | 0 | |
| 2 | USA (Pennsylvania, serologic evidence of infection in 4 animal handlers)[24]. | 1990 | 0 (0) | 0 | ||
| 3 | Philippines (Ebola-like virus present at primates export facilities)[25]. | 1989/90 | 0 (0) | 0 | ||
| 4 | Italy (Ebola-like virus causing hemorrhagic fever in Macaques imported from Philippines)[26]. | 1992 | 0 (0) | 0 | ||
| 5 | USA (Outbreak in a Texas quarantine facility due to infected cynomolgus macaques imported from Philippines. Human seroconversion was not detected)[27]. | 1996 | 0 (0) | 0 | ||
| 6 | Philippines (A single primate export facility in the island group of Luzon appeared to be the source of infected primates in the USA)[28]. | 1996 | 0 (0) | 0 | ||
| 7 | Philippines (outbreak occurred in two farms located in Bulacan & Pangasinan provinces. First report of a filovirus infecting a non-primate mammal)[29]. | 2009 | 6 (0) | 0 | ||
| Ebola-Ivory Coast | 1 | Cote-d'Ivoire, central west Africa (a 39-year-old female was infected when she autopsied a dead chimpanzee)[30]. | 1994 | 1 (0) | 0 | |
| Ebola-Bundibugyo | 1 | Uganda (outbreak occurred in Bundibugyo district, western Uganda. A new Ebola virus species was identified as the cause of the outbreak)[31]. | 2007/2008 (28 November to 20 February) | 149 (37) | 25 | |
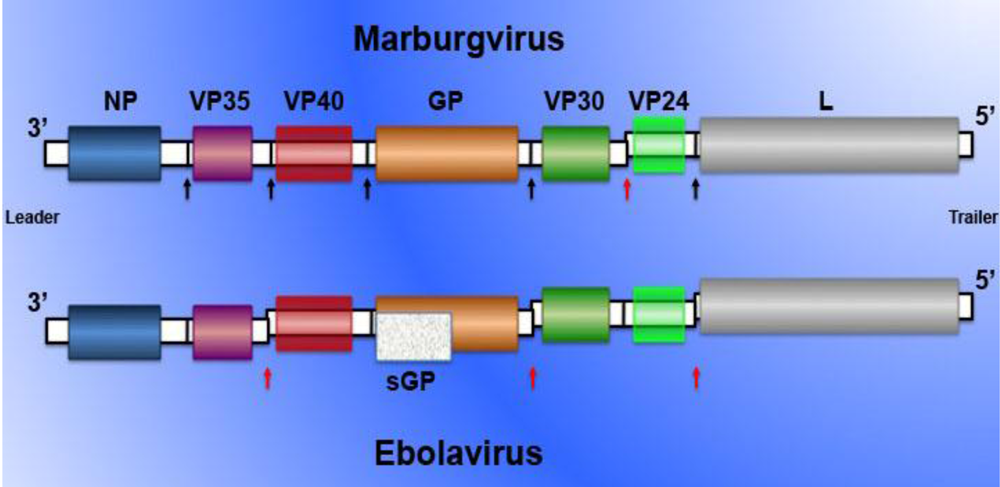
2. Filovirus pathogenesis
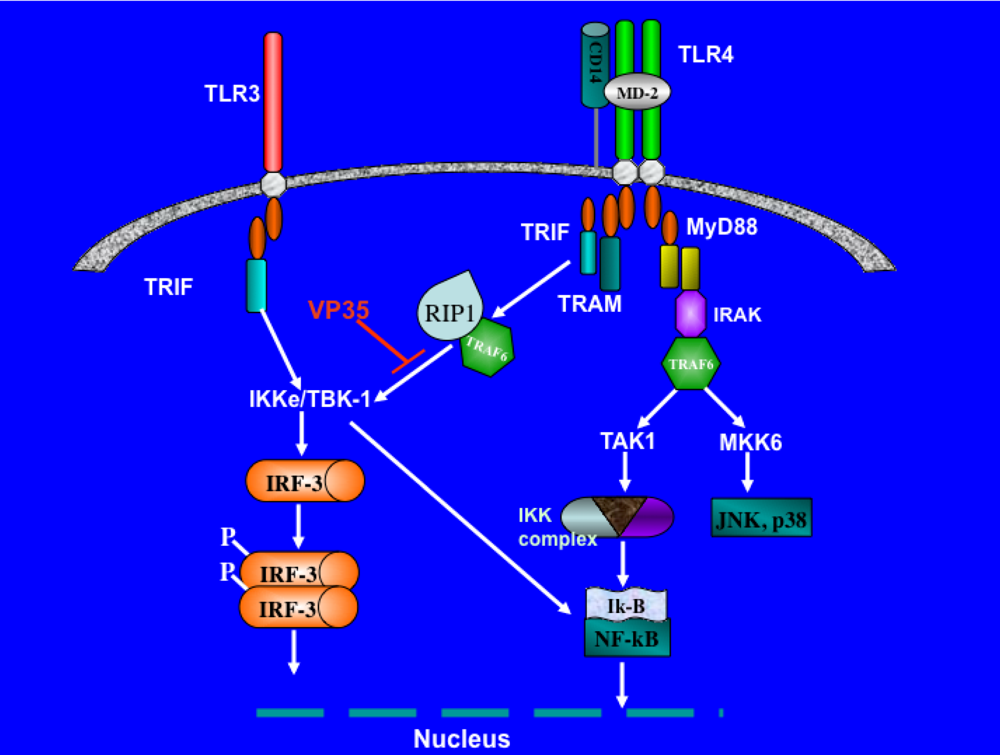
3. Evasion of interferon response by Filovirus
4. Conclusions
Acknowledgments
References
- Sanchez, A.; Geisbert, T.W.; Feldmann, H. Filoviridae: Marburg and Ebola viruses. Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1409–1448. [Google Scholar]
- Lamb, R.A. Mononegavirales. Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1357–1361. [Google Scholar]
- Kissling, R.E.; Robinson, R.Q.; Murphy, F.A.; Whitfield, S.G. Agent of disease contracted from green monkeys. Science 1968, 160, 888–890. [Google Scholar] [PubMed]
- World Health Organization. Ebola haemorrhagic fever in Zaire, 1976. Bull. World Health Org. 1978, 56, 271–293. [Google Scholar] [PubMed]
- World Health Organization. Ebola haemorrhagic fever in Sudan, 1976. Bull. World Health Org. 1978, 56, 247–270. [Google Scholar] [PubMed]
- World Health Organization. Marburg haemorrhagic fever. Available online: http://www.who.int/mediacentre/factsheets/fs_marburg/en/index.html (accessed 3 January 2010). [PubMed]
- Gear, J.S.; Cassel, G.A.; Gear, A.J.; Trappler, B.; Clausen, L.; Meyers, A.M.; Kew, M.C.; Bothwell, T.H.; Sher, R.; Miller, G.B.; Schneider, J.; Koornhof, H.J.; Gomperts, E.D.; Isaacson, M.; Gear, J.H. Outbreake of Marburg virus disease in Johannesburg. Br. Med. J. 1975, 4, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.G.; Nichol, S.T.; Muyembe-Tamfum, J.J.; Borchert, M.; Rollin, P.E.; Sleurs, H.; Campbell, P.; Tshioko, F.K.; Roth, C.; Colebunders, R.; Pirard, P.; Mardel, S.; Olinda, L.A.; Zeller, H.; Tshomba, A.; Kulidri, A.; Libande, M.L.; Mulangu, S.; Formenty, P.; Grein, T.; Leirs, H.; Braack, L.; Ksiazek, T.; Zaki, S.; Bowen, M.D.; Smit, S.B.; Leman, P.A.; Burt, F.J.; Kemp, A.; Swanepoel, R. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N. Engl. J. Med. 2006, 355, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Stroher, U.; Gomes da Silva, F.; del Castillo, F.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [PubMed]
- Center for Disease Control and Prevention. Known cases and outbreaks of Marburg hemorrhagic fever, in chronological order. Available online: http://www.cdc.gov/ncidod/dvrd/spb/mnpages/dispages/marburg/marburgtable.htm (accessed 3 January 2010). [CrossRef] [PubMed]
- Heymann, D.L.; Weisfeld, J.S.; Webb, P.A.; Johnson, K.M.; Cairns, T.; Berquist, H. Ebola hemorrhagic fever: Tandala, Zaire, 1977-178. J. Infect. Dis. 1980, 142, 372–376. [Google Scholar] [PubMed]
- Georges, A.J.; Leroy, E.M.; Renaut, A.A.; Benissan, C.T.; Nabias, R.J.; Ngoc, M.T.; Obiang, P.I.; Lepage, J.P.; Bertherat, E.J.; Bénoni, D.D.; Wickings, E.J.; Amblard, J.P.; Lansoud-Soukate, J.M.; Milleliri, J.M.; Baize, S.; Georges-Courbot, M.C. Ebola hemorrhagic fever outbreak in Gabon, 1994-1997: epidemiologic and health control issues. J. Infect. Dis. 1999, 179, S65–S75. [Google Scholar] [PubMed]
- Khan, A.S.; Tshioko, K.; Heymann, D.L.; Le Guenno, B.; Nabeth, P.; Kerstiëns, B.; Fleerackers, Y.; Kilmarx, P.H.; Rodier, G.R.; Nkuku, O.; Rollin, P.E.; Sanchez, A.; Zaki, S.R.; Swanepoel, R.; Tomori, O.; Nichol, S.T.; Peters, C.J.; Muyembe-Tamfum, J.J.; Ksiazek, T.G. The reemergence of ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999, 179, S76–S86. [Google Scholar] [PubMed]
- World Health Organization. Ebola haemorrhagic fever--South Africa. Weekly Epidemiol. Rec. 1996, 71, 359. [Google Scholar]
- Public Health Agency of Canada. Outbreak of Ebola hemorrhagic fever, Congo and Gabon, October 2001 to July 2002. Can. Comm. Dis. Rep. 2003, 29, 15. [Google Scholar]
- World Health Organization. Outbreaks of Ebola haemorrhagic fever in the Republic of the Congo, January-April 2003. Weekly Epidemiol. Rec. 2003, 78, 285–289. [Google Scholar]
- World Health Organization. Ebola haemorrhagic fever, Republic of the Congo-update. Weekly Epidemiol. Rec. 2003, 78, 409. [Google Scholar]
- World Health Organization Global Alert and Response (GAR). Ebola. Available online: http://www.who.int/csr/don/archive/disease/ebola_haemorrhagic_fever/en/ (accessed 4 January 2010).
- Emond, R.T.; Evans, B.; Bowen, E.T.; Lloyd, G. A case of Ebola virus infection. Br. Med. J. 1977, 2, 541–544. [Google Scholar] [CrossRef]
- Baron, R.C.; McCormick, J.B.; Zubeir, O.A. Ebola virus disease in southern Sudan: hospital dissemination and intrafamilial spread. Bull. World Health Org. 1983, 61, 997–1003. [Google Scholar] [PubMed]
- Okware, S.I.; Omaswa, F.G.; Zaramba, S.; Opio, A.; Lutwama, J.J.; Kamugisha, J.; Rwaguma, E.B.; Kagwa, P.; Lamunu, M. An outbreak of Ebola in Uganda. Trop. Med. Int. Health 2002, 7, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Outbreak of Ebola haemorrhagic fever in Yambio, south Sudan, April-June 2004. Weekly Epidemiol. Rec. 2005, 80, 370–375. [Google Scholar]
- Jahrling, P.B.; Geisbert, T.W.; Dalgard, D.W.; Johnson, E.D.; Ksiazek, T.G.; Hall, W.C.; Peters, C.J. Preliminary report: isolation of Ebola virus from monkey imported to USA. Lancet 1990, 335, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Groseth, A.; Ströher, U.; Theriault, S.; Feldmann, H. Molecular characterization of an isolate from the 1989/90 epizootic of Ebola virus Reston among macaques imported into the United States. Virus Res. 2002, 87, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.G.; Burans, J.P.; Ksiazek, T.G.; Del Rosario, R.A.; Miranda, M.E.; Manaloto, C.R.; Barrientos, A.B.; Robles, C.G.; Dayrit, M.M.; Peters, C.J. Outbreak of fatal illness among captive macaques in the Philippines caused by an ebola-related filovirus. Am. J. Trop. Med. Hyg. 1992, 46, 664–671. [Google Scholar] [PubMed]
- World Health Organization. Viral haemorrhagic fever in imported monkeys. Weekly Epidemiol. Rec. 1992, 67, 142. [Google Scholar]
- Rollin, P.E.; Williams, R.J.; Bressler, D.S.; Pearson, S.; Cottingham, M.; Pucak, G.; Sanchez, A.; Trappier, S.G.; Peters, R.L.; Greer, P.W.; Zaki, S.R.; Demarcus, T.; Hendricks, K.; Kelley, M.; Simpson, D.; Geisbert, T.W.; Jahrling, P.B.; Peters, C.J.; Ksiazek, T.G. Ebola (subtype Reston) virus among quarantined nonhuman primates recently imported from the Philippines to the United States. J. Infect. Dis. 1999, 179, S108–S114. [Google Scholar] [PubMed]
- Miranda, M.E.; White, M.E.; Dayrit, M.M.; Hayes, C.G.; Ksiazek, T.G.; Burans, J.P. Seroepidemiological study of filovirus realted to Ebola in the Philippines. Lancet 1991, 337, 425–426. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Ebola Reston in pigs and humans, Philippines. Weekly Epidemiol. Rec. 2009, 84, 49–50. [Google Scholar]
- Le Guenno, B.; Formenty, P.; Wyers, M.; Gounon, P.; Walker, F.; Boesch, C. Isolation and partial charaterisation of a new strain of Ebola virus. Lancet 1995, 345, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Sealy, T.K.; Khristova, M.L.; Albarino, C.G.; Conlan, S.; Reeder, S.A.; Quan, P.-L.; Lipkin, W.I.; Downing, R.; Tappero, J.W.; Okware, S.; Lutwama, J.; Bakamutumaho, B.; Kayiwa, J.; Comer, J.A.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008, 4, e1000212. [Google Scholar] [CrossRef] [PubMed]
- Formenty, P.; Hatz, C.; Le Guenno, B.; Stoll, A.; Rogenmoser, P.; Widmer, A. Human infection due to Ebola virus, subtype Cote d'Ivoire: clinical and biologic presentation. J. Infect. Dis. 1999, 179, S48–S3. [Google Scholar] [PubMed]
- Breman, J.G.; Johnson, K.M.; van der Groen, G.; Robbins, C.B.; Szczeniowski, M.V.; Ruti, K.; Webb, P.A.; Meier, F.; Heymann, D.L. A search for Ebola virus in animals in the Democratic Republic of the Congo and Cameroon: ecologic, virologic, and serologic surveys, 1979-1980. Ebola Virus Study Teams. J. Infect. Dis. 1999, 179, S139–S147. [Google Scholar] [PubMed]
- Towner, J.S.; Pourrut, X.; Albarino, C.G.; Nkogue, C.N.; Bird, B.H.; Grard, G.; Ksiazek, T.G.; Gonzalez, J.-P.; Nichol, S.T.; Leroy, E.M. Marburg virus infection detected in a common African bat. PLoS One 2007, 2, e764. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Swanepoel, R.; Leman, P.A.; Burt, F.J.; Zachariades, N.A.; Braack, L.E.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Peters, C.J. Experimental inoculation of plants and animals with Ebola virus. Emerg. Infect. Dis. 1996, 2, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.R.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; Formenty, P.B.H.; Albarino, C.G.; Miller, D.M.; Reed, Z.D.; Kayiwa, J.T.; Mills, J.N.; Cannon, D.L.; Greer, P.W.; Byaruhanga, E.; Farnon, E.C.; Atimnedi, P.; Okware, S.; Katongole-Mbidde, E.; Downing, R.; Tappero, J.W.; Zaki, S.R.; Ksiazek, T.G.; Nichol, S.T.; Rollin, P.E. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e100000536. [Google Scholar] [CrossRef]
- Barrette, R.W.; Metwally, S.A.; Rowland, J.M.; Xu, L.; Zaki, S.R.; Nichol, S.T.; Rollin, P.E.; Towner, J.S.; Shieh, W.-J.; Batten, B.; Sealy, T.K.; Carrillo, C.; Moran, K.E.; Bracht, A.J.; Mayr, G.A.; Sirios-Cruz, M.; Catbagan, D.P.; Lautner, E.A.; Ksiazek, T.G.; White, W.R.; McIntosh, M.T. Discovery of swine as a host for the Reston ebolavirus. Science 2009, 325, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Mahanty, S.; Bray, M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect. Dis. 2004, 4, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Jahrling, P.B. Exotic emerging viral diseases: progress and challenges. Nat. Med. 2004, 10, S110–S121. [Google Scholar] [CrossRef] [PubMed]
- Bwaka, M.A.; Bonnet, M.J.; Calain, P.; Colebunders, R.; De Roo, A.; Guimard, Y.; Katwiki, K.R.; Kibadi, K.; Kipasa, M.A.; Kuvula, K.J.; Mapanda, B.B.; Massamba, M.; Mupapa, K.D.; Muyembe-Tamfum, J.J.; Ndaberey, E.; Peters, C.J.; Rollin, P.E.; Van den Enden, E.; Van den Enden, E. Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observations in 103 patients. J. Infect. Dis. 1999, 179, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Villinger, F.; Rollin, P.E.; Brar, S.S.; Chikkala, N.F.; Winter, J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R.; Ansari, A.A.; Peters, C.J. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis. 1999, 179, S188–S191. [Google Scholar] [PubMed]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.C.; Capron, M.; Lansoud-Soukate, J.M.; Debre, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.-C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.M.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Baize, S.; Volchkov, V.E.; Fisher-Hoch, S.P.; Georges-Courbot, M.C.; Lansoud-Soukate, J.M.; Capron, M.; Debre, P.; McCormick, J.B.; Georges, A.J. Human asymptomatic Ebola infection and strong inflammatory response. Lancet 2000, 355, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Feldmann, H.; Stroher, U.; Geisbert, J.B.; Fernando, L.; Grolla, A.; Klenk, H.D.; Sullivan, N.J.; Volchkov, V.E.; Fritz, E.A.; Daddario, K.M.; Hensley, L.E.; Jahrling, P.B.; Geisbert, T.W. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005, 11, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Geisbert, T.W.; Geisbert, J.B.; Shedlock, D.J.; Xu, L.; Lamoreaux, L.; Custers, J.H.H.V.; Popernack, P.M.; Yang, Z.-Y.; Pau, M.G.; Roederer, M.; Koup, R.A.; Goudsmit, J.; Jahrling, P.B.; Nabel, G.J. Immune Protection of Nonhuman Primates against Ebola Virus with Single Low-Dose Adenovirus Vectors Encoding Modified GPs. PLoS Med. 2006, 3, e177. [Google Scholar] [CrossRef] [PubMed]
- Oswald, W.B.; Geisbert, T.W.; Davis, K.J.; Geisbert, J.B.; Sullivan, N.J.; Jahrling, P.B.; Parren, P.W.; Burton, D.R. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. 2007, 3, e9. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Geisbert, T.W.; Geisbert, J.B.; Swearengen, J.R.; Bray, M.; Jaax, N.K.; Huggins, J.W.; LeDuc, J.W.; Peters, C.J. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 1999, 179, S224–S234. [Google Scholar] [PubMed]
- Parren, P.W.H.I.; Geisbert, T.W.; Maruyama, T.; Jahrling, P.B.; Burton, D.R. Pre- and postexposure prophylaxis of Ebola virus infection in an animal model by passive transfer of a neutralizing human antibody. J. Virol. 2002, 76, 6408–6412. [Google Scholar] [CrossRef] [PubMed]
- Kudoyarova-Zubavichene, N.M.; Sergeyev, N.N.; Chepurnov, A.A.; Netesov, S.V. Preparation and use of hyperimmune serum for prophylaxis and therapy of Ebola virus infections. J. Infect. Dis. 1999, 179, S218–S223. [Google Scholar] [PubMed]
- Mupapa, K.; Massamba, M.; Kibadi, K.; Kuvula, K.; Bwaka, A.; Kipasa, M.; Colebunders, R.; Muyembe-Tamfum, J.J. Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. J. Infect. Dis. 1999, 179, S18–S23. [Google Scholar] [PubMed]
- Jahrling, P.B.; Geisbert, J.B.; Swearengen, J.R.; Larsen, T.; Geisbert, T.W. Ebola hemorrhagic fever: evaluation of passive immunotherapy in nonhuman primates. J. Infect. Dis. 2007, 196, S400–S403. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Baize, S.; Debre, P.; Lansoud-Soukate, J.M.; Mavoungou, E. Early immune responses accompanying human asymptomatic Ebola infections. Clin. Exp. Immunol. 2001, 124, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Bosio, C.M.; Aman, M.J.; Grogan, C.; Hogan, R.; Ruthel, G.; Negley, D.; Mohamadzadeh, M.; Bavari, S.; Schmaljohn, A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J. Infect. Dis. 2003, 188, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [PubMed]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; McRae, M.; Rollin, P.E.; Pulendran, B. Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar] [PubMed]
- Bradfute, S.B.; Warfield, K.L.; Bavari, S. Functional CD8+ T cell responses in lethal ebola virus infection. J. Immnunol. 2008, 180, 4058–4066. [Google Scholar]
- Hermiston, M.L.; Xu, Z.; Weiss, A. CD45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003, 21, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Bradfute, S.B.; Peyser, B.D.; Warfield, K.L.; Ruthel, G.; Lane, D.; Kenny, T.A.; Anderson, A.O.; Raschke, W.C.; Bavari, S. Reduced levels of protein tyrosine phosphatase CD45 protect mice from the lethal effects of Ebola virus infection. Cell Host Microbe 2009, 6, 162–173. [Google Scholar] [CrossRef]
- Warfield, K.L.; Perkins, J.G.; Swenson, D.L.; Deal, E.M.; Bosio, C.M.; Aman, M.J.; Yokoyama, W.M.; Young, H.A.; Bavari, S. Role of natural killer cells in innate protection against lethal ebola virus infection. J. Exp. Med. 2004, 200, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.L.; Ruthel, G.; Warfield, K.L.; Swenson, D.L.; Bosio, C.M.; Aman, M.J.; Bavari, S. NKp30-dependent cytolysis of filovirus-infected human dendritic cells. Cell. Microbiol. 2007, 9, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Mohamadzadeh, M.; Chen, L.; Schmaljohn, A.L. How Ebola and Marburg viruses battle the immune system. Nat. Rev. Immunol. 2007, 7, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Volchkova, V.A.; Muhlberger, E.; Kolesnikova, L.V.; Weik, M.; Dolnik, O.; Klenk, H.D. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science 2001, 291, 1965–1969. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar] [CrossRef]
- Chan, S.Y.; Ma, M.C.; Goldsmith, M.A. Differential induction of cellular detachment by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Gen. Virol. 2000, 81, 2155–2159. [Google Scholar] [PubMed]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Kagan, E.; Hensley, L.E. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003, 188, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Hensley, L.E.; Jahrling, P.B.; Larsen, T.; Geisbert, J.B.; Paragas, J.; Young, H.A.; Fredeking, T.M.; Rote, W.E.; Vlasuk, G.P. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 2003, 362, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Weber, F. Pathogenic viruses: smart manipulators of the interferon system. Curr. Top. Microbiol. Immunol. 2007, 316, 315–334. [Google Scholar] [PubMed]
- Malmgaard, L. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 2004, 24, 439–454. [Google Scholar] [PubMed]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Sanchez, A.; Offermann, M.K. Ebola virus inhibits induction of genes by double-stranded RNA in endothelial cells. Virology 1998, 252, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Sanchez, A.; Offermann, M.K. Ebola virus selectively inhibits responses to interferons, but not to interleukin-1beta, in endothelial cells. J. Virol. 1999, 73, 3491–3496. [Google Scholar] [PubMed]
- Gupta, M.; Mahanty, S.; Ahmed, R.; Rollin, P.E. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with ebola virus secrete MIP-1alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology 2001, 284, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Bray, M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001, 82, 1365–1373. [Google Scholar] [PubMed]
- Basler, C.F.; Mikulasova, A.; Martinez-Sobrido, L.; Paragas, J.; Muhlberger, E.; Bray, M.; Klenk, H.D.; Palese, P.; Garcia-Sastre, A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003, 77, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Wang, X.; Muhlberger, E.; Volchkov, V.; Paragas, J.; Klenk, H.D.; Garcia-Sastre, A.; Palese, P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA 2000, 97, 12289–12294. [Google Scholar] [CrossRef]
- Chang, T.-H.; Kubota, T.; Matsuoka, M.; Jones, S.; Bradfute, S.B.; Bray, M.; Ozato, K. Ebola Zaire Virus Blocks Type I Interferon Production by Exploiting the Host SUMO Modification Machinery. PLoS Pathog. 2009, 5, e1000493. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.; Smith, E.; Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [PubMed]
- Barnes, B.; Lubyova, B.; Pitha, P.M. On the role of IRF in host defense. J. Interferon Cytokine Res. 2002, 22, 59–71. [Google Scholar] [PubMed]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [PubMed]
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661. [Google Scholar] [CrossRef]
- Izaguirre, A.; Barnes, B.J.; Amrute, S.; Yeow, W.-S.; Megjugorac, N.; Dai, J.; Feng, D.; Chung, E.; Pitha, P.M.; Fitzgerald-Bocarsly, P. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 2003, 74, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Grandvaux, N.; Sharma, S.; Tenoever, B.R.; Servant, M.J.; Lin, R. Convergence of the NF-kappaB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238. [Google Scholar] [CrossRef]
- Uematsu, S.; Sato, S.; Yamamoto, M.; Hirotani, T.; Kato, H.; Takeshita, F.; Matsuda, M.; Coban, C.; Ishii, K.J.; Kawai, T.; Takeuchi, O.; Akira, S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 2005, 201, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Cardenas, W.B.; Basler, C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kawai, T.; Takeda, K.; Matsumoto, M.; Inoue, J.; Tatsumi, Y.; Kanamaru, A.; Akira, S. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 1999, 11, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.T.; Maniatis, T. A new family of IKK-related kinases may function as I kappa B kinase kinases. Biochim. Biophys. Acta. 2001, 1471, M57–62. [Google Scholar] [PubMed]
- Perry, A.K.; Chow, E.K.; Goodnough, J.B.; Yeh, W.C.; Cheng, G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 2004, 199, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The Roles of Two IkappaB Kinase-related Kinases in Lipopolysaccharide and Double Stranded RNA Signaling and Viral Infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Geisbert, T.W. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005, 37, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yan, Z.; Prabhakar, B.; Feng, Z.; Ma, Y.; Verpooten, D.; Ganesh, B.; He, B. The VP35 Protein of Ebola Virus Impairs Dendritic Cell Maturation Induced by Virus and Lipopolysaccharide (Epub October 2009). J. Gen. Virol. 2009. [Google Scholar]
- Cardenas, W.B.; Loo, Y.M.; Gale Jr., M.; Hartman, A.L.; Kimberlin, C.R.; Martinez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 2004, 328, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Dover, J.E.; Towner, J.S.; Nichol, S.T. Reverse genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J. Virol. 2006, 80, 6430–6440. [Google Scholar] [CrossRef] [PubMed]
- Schumann, M.; Gantke, T.; Muhlberger, E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J. Virol. 2009, 83, 8993–8997. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Cerveny, M.; Yan, Z.; He, B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007, 81, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; de Vries, W.; Geutjes, E.-J.; Prins, M.; de Haan, P.; Berkhout, B. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Ginder, N.D.; Fulton, D.B.; Nix, J.; Basler, C.F.; Honzatko, R.B.; Amarasinghe, G.K. Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. USA 2009, 106, 411–416. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale Jr., M.; Akira, S.; Yonehara, S.; Kato, A.; Fujita, T. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA Is an Adapter Protein Required for Virus-Triggered IFN-beta Signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.; Vaidya, S.; O'Connell, R.; Dadgostar, H.; Dempsey, P.; Wu, T.; Rao, G.; Sun, R.; Haberland, M.; Modlin, R.; Cheng, G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 2002, 17, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; Freudenberg, M.; Ricciardi-Castagnoli, P.; Layton, B.; Beutler, B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, L.A.; Fitzgerald, K.A.; Bowie, A.G. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003, 24, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; Akira, S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Uematsu, S.; Hoshino, K.; Kaisho, T.; Takeuchi, O.; Takeda, K.; Akira, S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 2003, 4, 1144–1150. [Google Scholar] [CrossRef]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; Crozat, K.; Sovath, S.; Han, J.; Beutler, B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Pitha, P.; Yoshimura, A.; Harty, R. Interaction Between Ebola Virus Glycoprotein and Host TLR-4 Leads to Induction of Pro-Inflammatory Cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Prele, C.M.; Woodward, E.A.; Bisley, J.; Keith-Magee, A.; Nicholson, S.E.; Hart, P.H. SOCS1 regulates the IFN but not NFkappaB pathway in TLR-stimulated human monocytes and macrophages. J. Immunol. 2008, 181, 8018–8026. [Google Scholar] [PubMed]
- Martinez, O.; Valmas, C.; Basler, C.F. Ebola virus-like particle-induced activation of NF-kappaB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 2007, 364, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Conzelmann, K.K. Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. J. Virol. 2005, 79, 5241–5247. [Google Scholar] [CrossRef] [PubMed]
- Mohamadzadeh, M.; Chen, L.; Schmaljohn, A.L. How ebola and marburg viruses battle the immune system. Nat. Rev. Immunol. 2007, 7, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Leung, L.W.; Hartman, A.L.; Martinez, O.; Shaw, M.L.; Carbonnelle, C.; Volchkov, V.E.; Nichol, S.T.; Basler, C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 2006, 80, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Valmas, C.; Martinez, O.; Sanchez, F.M.; Basler, C.F. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 2007, 81, 13469–13477. [Google Scholar] [CrossRef] [PubMed]
- Mateo, M.; Reid, S.P.; Leung, L.W.; Basler, C.F.; Volchkov, V.E. Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signalling. J. Virol. 2010, 84, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Mühlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.; Katze, M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Cárdenas, W.B. Evasion of the Interferon-Mediated Antiviral Response by Filoviruses. Viruses 2010, 2, 262-282. https://doi.org/10.3390/v2010262
Cárdenas WB. Evasion of the Interferon-Mediated Antiviral Response by Filoviruses. Viruses. 2010; 2(1):262-282. https://doi.org/10.3390/v2010262
Chicago/Turabian StyleCárdenas, Washington B. 2010. "Evasion of the Interferon-Mediated Antiviral Response by Filoviruses" Viruses 2, no. 1: 262-282. https://doi.org/10.3390/v2010262
APA StyleCárdenas, W. B. (2010). Evasion of the Interferon-Mediated Antiviral Response by Filoviruses. Viruses, 2(1), 262-282. https://doi.org/10.3390/v2010262