
Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.2. Mutational Scanning of the Binding Interfaces for the SARS-CoV-2 S Protein Complexes with Antibodies
2.3. Binding Free Energy Computations of the SARS-CoV-2 S Protein Complexes with Antibodies
3. Results
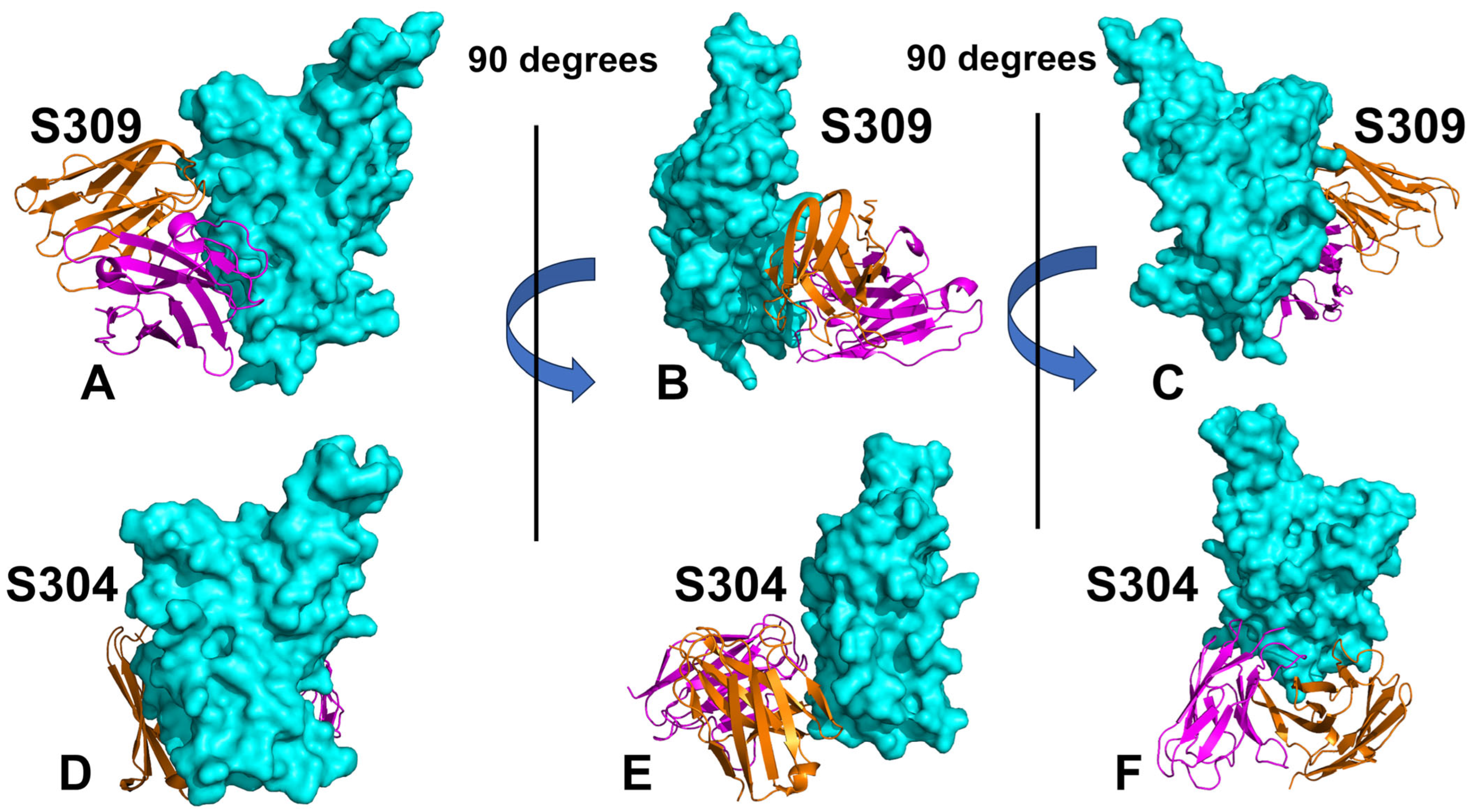
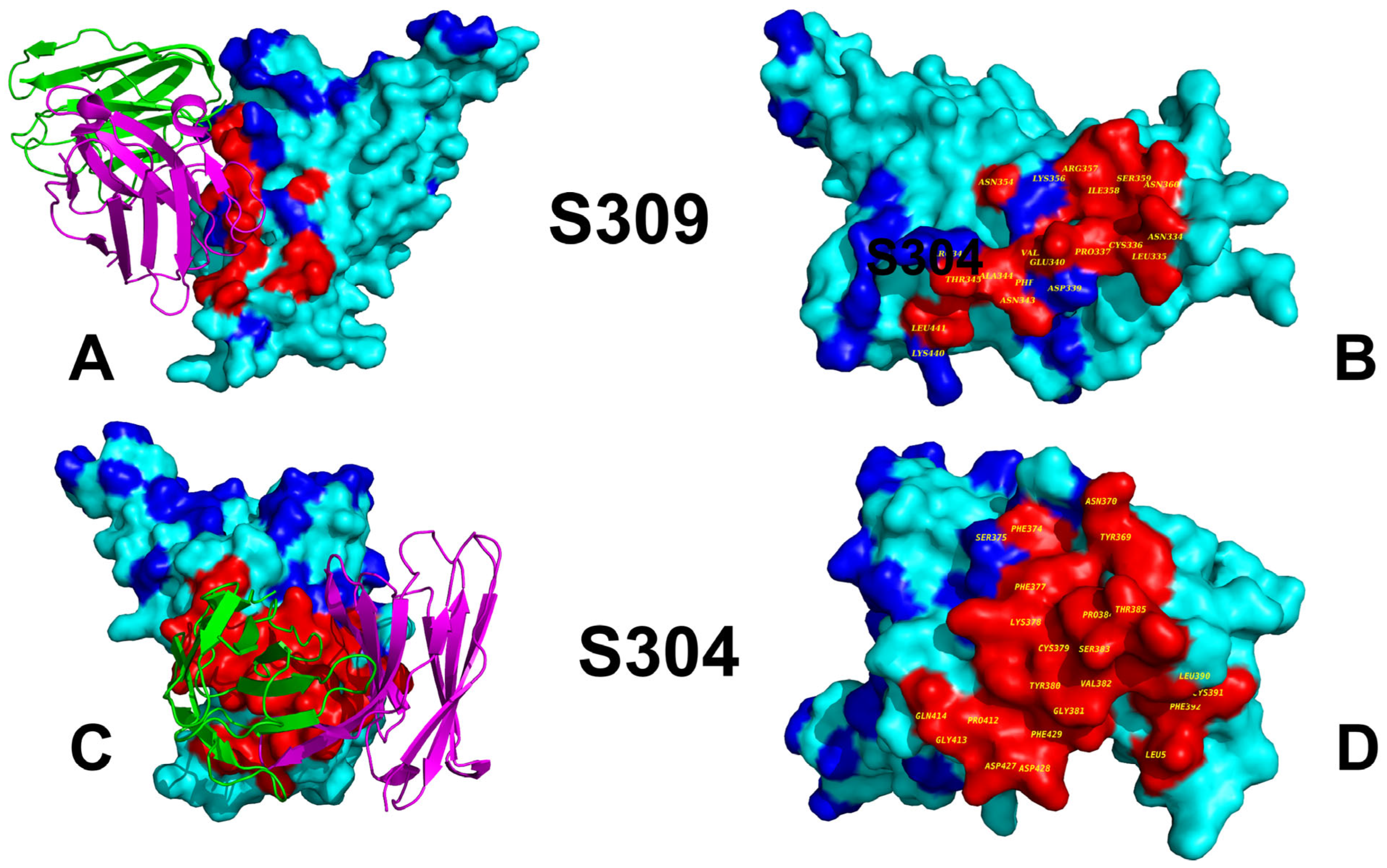
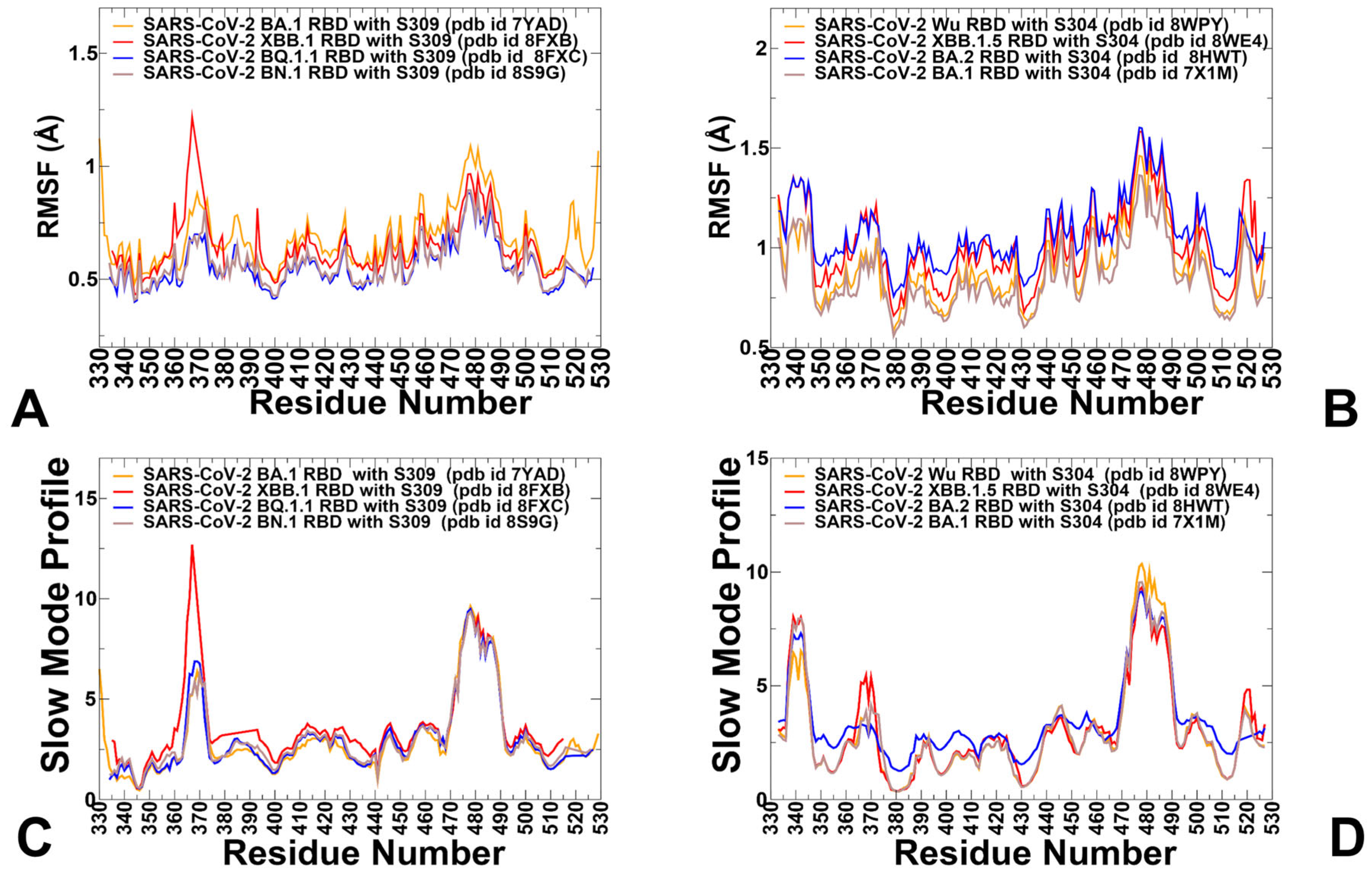
3.1. Structural Analysis and MD Simulations of the S-RBD Complexes with Antibodies
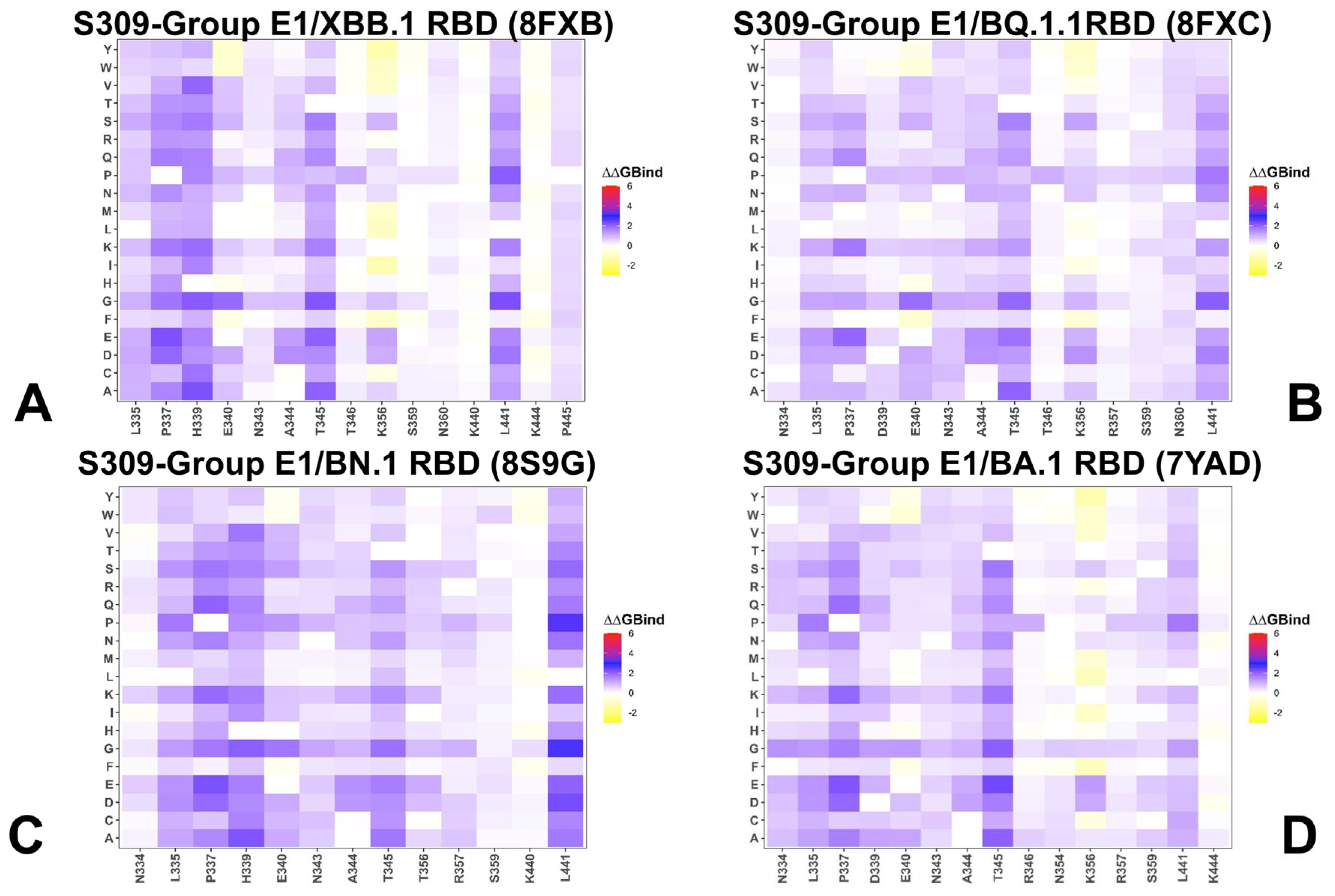
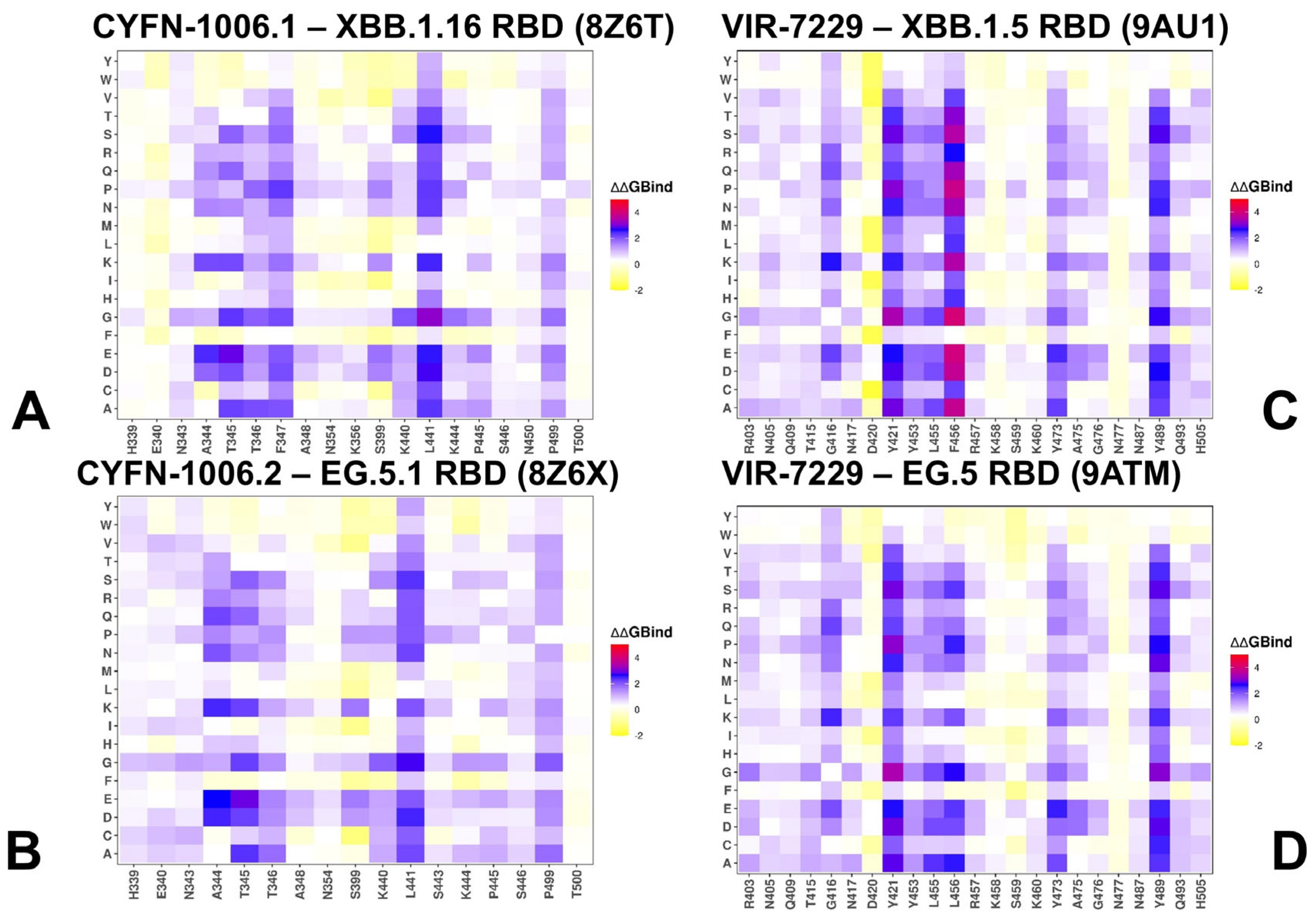
3.2. Mutational Profiling of Protein Binding Interfaces
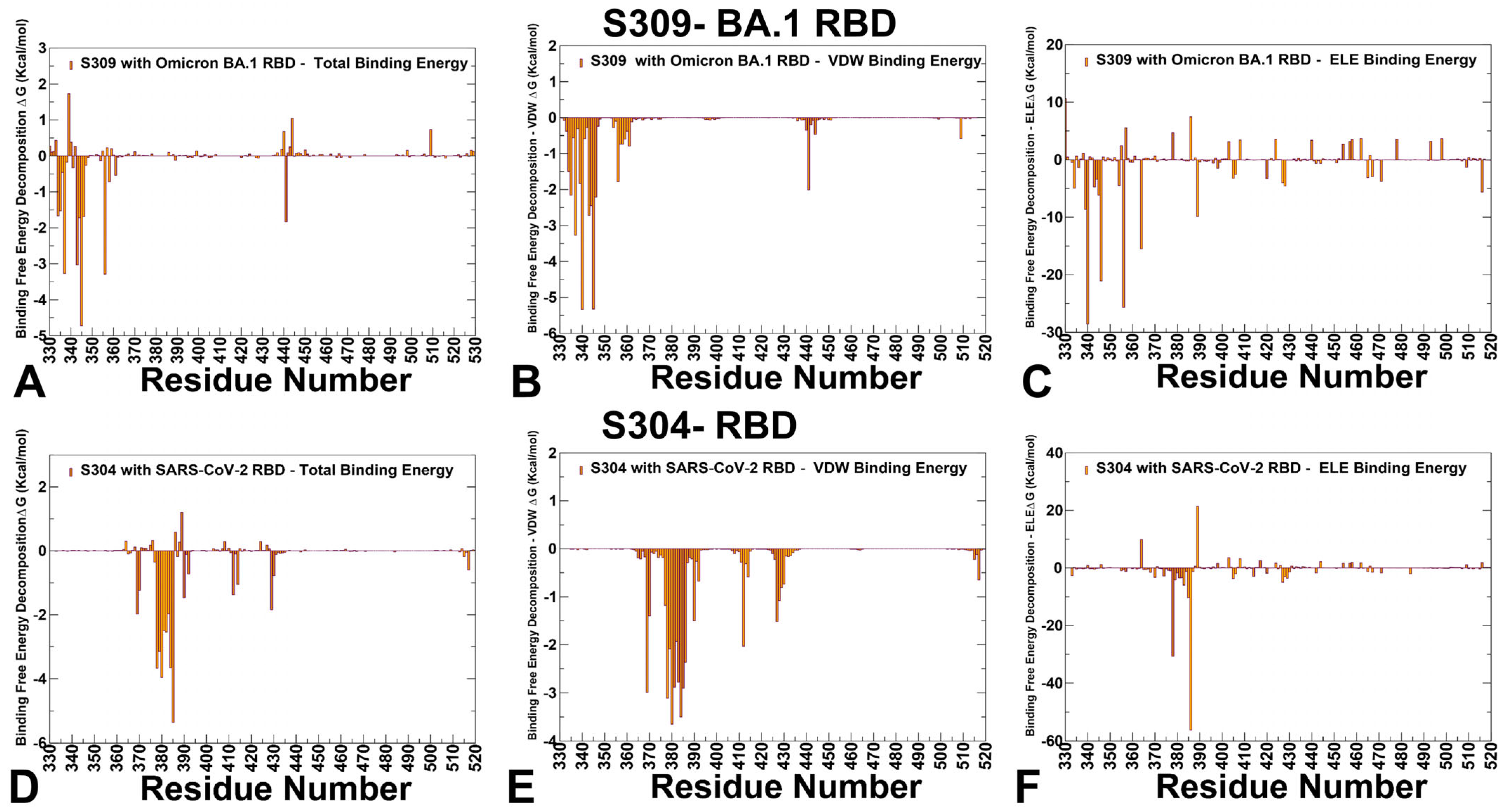
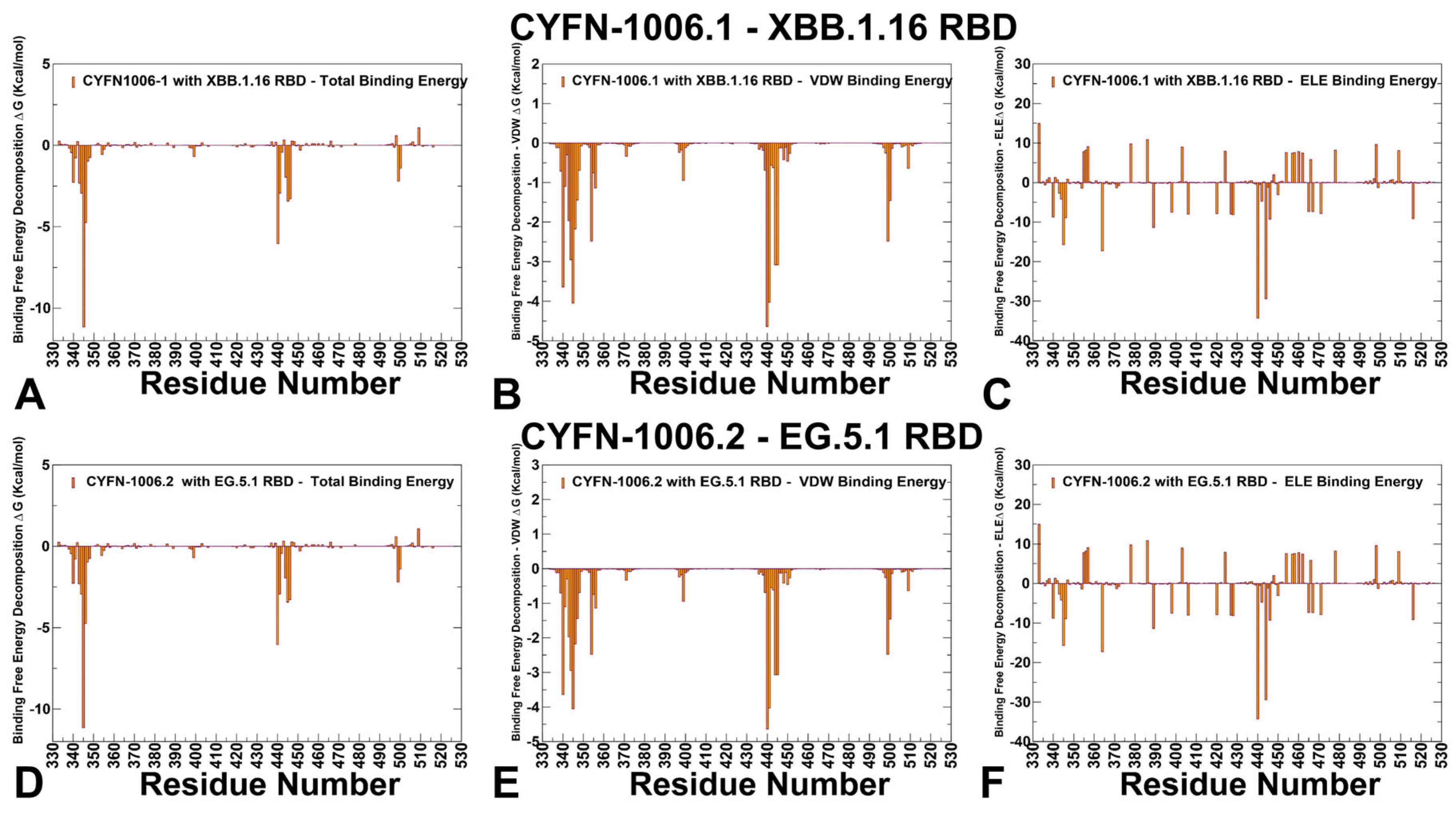
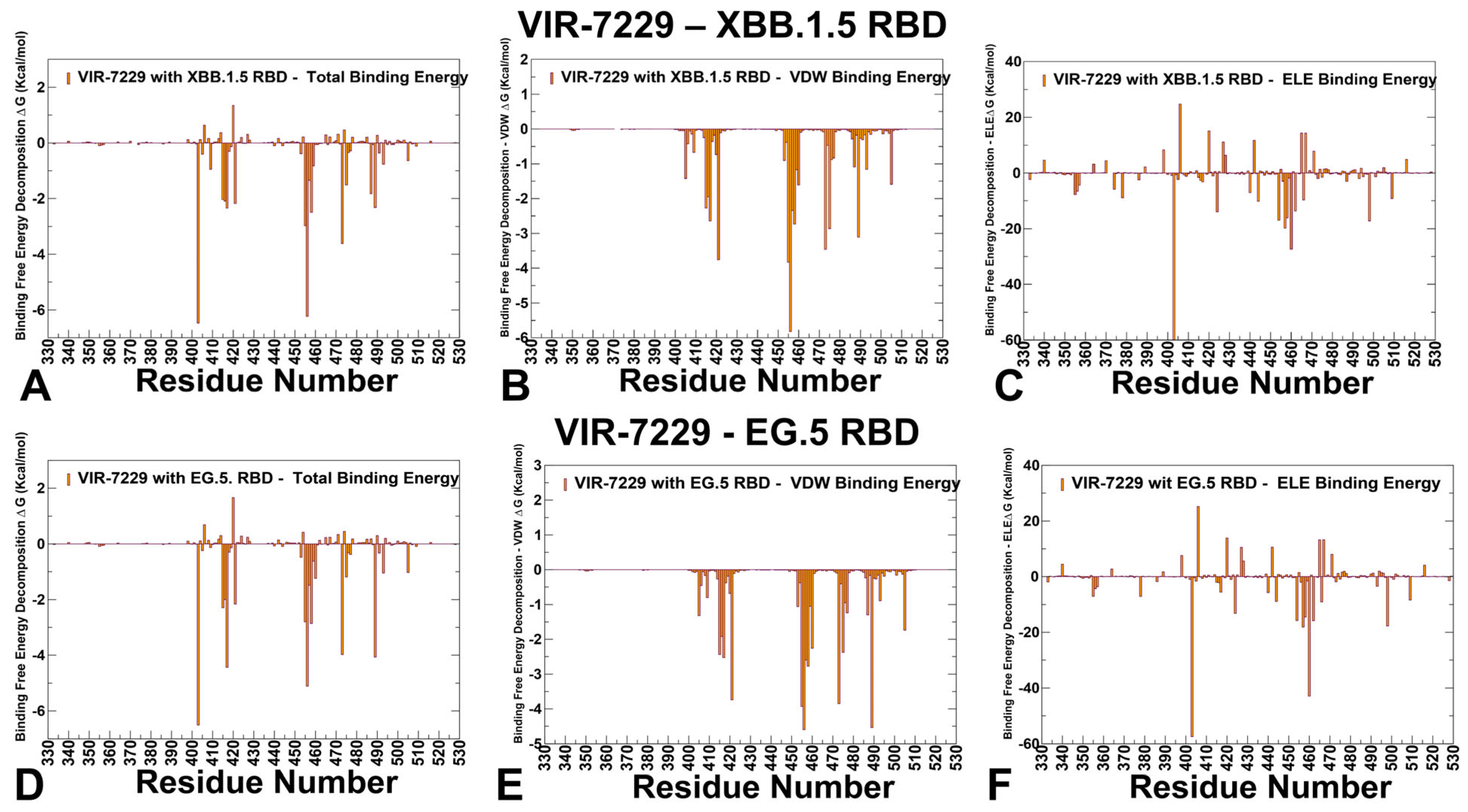
3.3. MM-GBSA Analysis of the Binding Affinities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes. Nat. Struct. Mol. Biol. 2022, 27, 229–238. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, C.; Wang, Y.; Hong, Q.; Zhang, C.; Li, Z.; Xu, S.; Zuo, Q.; Liu, C.; Huang, Z.; et al. Conformational dynamics of the Beta and Kappa SARS-CoV-2 spike proteins and their complexes with ACE2 receptor revealed by cryo-EM. Nat Commun. 2021, 12, 7345. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schuerman, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. eLife 2022, 11, e75433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron Variant: AntibodyEvasion and Cryo-EM Structure of Spike Protein–ACE2 Complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular Basis of Receptor Binding and AntibodyNeutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Calvaresi, V.; Wrobel, A.G.; Toporowska, J.; Hammerschmid, D.; Doores, K.J.; Bradshaw, R.T.; Parsons, R.B.; Benton, D.J.; Roustan, C.; Reading, E.; et al. Structural dynamics in the evolution of SARS-CoV-2 spike glycoprotein. Nat. Commun. 2023, 14, 1421. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike Trimer with ACE2 and an Anti-Omicron Ab. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural Diversity of the SARS-CoV-2 Omicron Spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.-J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-Neutralization of SARS-CoV-2 by a Human Monoclonal SARS-CoV Antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent Human Antibodies Protect against SARS-CoV-2 Challenge via Multiple Mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, H.; Wu, N.C.; Lee, C.-C.D.; Zhu, X.; Zhao, F.; Huang, D.; Yu, W.; Hua, Y.; Tien, H.; et al. Structural Basis of a Shared Antibody Response to SARS-CoV-2. Science 2020, 369, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Piccoli, L.; Case, J.B.; Park, Y.-J.; Beltramello, M.; Guarino, B.; Dang, H.; de Melo, G.D.; Pinto, D.; Sprouse, K.; et al. Neutralization, Effector Function and Immune Imprinting of Omicron Variants. Nature 2023, 621, 592–601. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural Basis of SARS-CoV-2 Omicron Immune Evasion and Receptor Engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Magnus, C.L.; Hiergeist, A.; Schuster, P.; Rohrhofer, A.; Medenbach, J.; Gessner, A.; Peterhoff, D.; Schmidt, B. Targeted Escape of SARS-CoV-2 in Vitro from Monoclonal Antibody S309, the Precursor of Sotrovimab. Front. Immunol. 2022, 13, 966236. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.-W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody Evasion Properties of SARS-CoV-2 Omicron Sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.-W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Huang, M.; Wu, L.; Zheng, A.; Xie, Y.; He, Q.; Rong, X.; Han, P.; Du, P.; Han, P.; Zhang, Z.; et al. Atlas of Currently Available Human Neutralizing Antibodies against SARS-CoV-2 and Escape by Omicron Sub-Variants BA.1/BA.1.1/BA.2/BA.3. Immunity 2022, 55, 1501–1514.e3. [Google Scholar] [CrossRef]
- Cui, L.; Li, T.; Xue, W.; Zhang, S.; Wang, H.; Liu, H.; Gu, Y.; Xia, N.; Li, S. Comprehensive Overview of Broadly Neutralizing Antibodies against SARS-CoV-2 Variants. Viruses 2024, 16, 900. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Han, P.; Zheng, A.; et al. Omicron SARS-CoV-2 Mutations Stabilize Spike up-RBD Conformation and Lead to a Non-RBM-Binding Monoclonal Antibody Escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef]
- Yu, L.; Wang, Y.; Liu, Y.; Xing, X.; Li, C.; Wang, X.; Shi, J.; Ma, W.; Li, J.; Chen, Y.; et al. Potent and Broadly Neutralizing Antibodies against Sarbecoviruses Elicited by Single Ancestral SARS-CoV-2 Infection. Commun. Biol. 2025, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.E.; Tortorici, M.A.; De Marco, A.; Pinto, D.; Foreman, W.B.; Taylor, A.L.; Park, Y.-J.; Bohan, D.; Rietz, T.; Errico, J.M.; et al. A Potent Pan-Sarbecovirus Neutralizing Antibody Resilient to Epitope Diversification. Cell 2024, 187, 7196–7213.e26. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Wec, A.Z.; Feng, L.; Yu, Y.; Wang, L.; Wang, P.; Yu, L.; Wang, J.; Hou, J.; Berrueta, D.M.; et al. A Generalized Framework to Identify SARS-CoV-2 Broadly Neutralizing Antibodies. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of interactions regulating conformational stability and dynamics of SARS-CoV-2 S-protein. Biophys. J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Xiao, S.; Alshahrani, M.; Gupta, G.; Tao, P.; Verkhivker, G. Markov State Models and Perturbation-Based Approaches Reveal Distinct Dynamic Signatures and Hidden Allosteric Pockets in the Emerging SARS-Cov-2 Spike Omicron Variant Complexes with the Host Receptor: The Interplay of Dynamics and Convergent Evolution Modulates Allostery and Functional Mechanisms. J. Chem. Inf. Model. 2023, 63, 5272–5296. [Google Scholar] [CrossRef]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P.; Verkhivker, G. AlphaFold2 Predictions of Conformational Ensembles and Atomistic Simulations of the SARS-CoV-2 Spike XBB Lineages Reveal Epistatic Couplings between Convergent Mutational Hotspots That Control ACE2 Affinity. J. Phys. Chem. B 2024, 128, 4696–4715. [Google Scholar] [CrossRef]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Verkhivker, G. Ensemble-Based Mutational Profiling and Network Analysis of the SARS-CoV-2 Spike Omicron XBB Lineages for Interactions with the ACE2 Receptor and Antibodies: Cooperation of Binding Hotspots in Mediating Epistatic Couplings Underlies Binding Mechanism and Immune Escape. Int. J. Mol. Sci. 2024, 25, 4281. [Google Scholar] [CrossRef]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Verkhivker, G. AlphaFold2 Modeling and Molecular Dynamics Simulations of the Conformational Ensembles for the SARS-CoV-2 Spike Omicron JN.1, KP.2 and KP.3 Variants: Mutational Profiling of Binding Energetics Reveals Epistatic Drivers of the ACE2 Affinity and Escape Hotspots of Antibody Resistance. Viruses 2024, 16, 1458. [Google Scholar] [CrossRef]
- Alshahrani, M.; Parikh, V.; Foley, B.; Raisinghani, N.; Verkhivker, G. Quantitative Characterization and Prediction of the Binding Determinants and Immune Escape Hotspots for Groups of Broadly Neutralizing Antibodies Against Omicron Variants: Atomistic Modeling of the SARS-CoV-2 Spike Complexes with Antibodies. Biomolecules 2025, 15, 249. [Google Scholar] [CrossRef]
- Alshahrani, M.; Parikh, V.; Foley, B.; Raisinghani, N.; Verkhivker, G. Mutational Scanning and Binding Free Energy Computations of the SARS-CoV-2 Spike Complexes with Distinct Groups of Neutralizing Antibodies: Energetic Drivers of Convergent Evolution of Binding Affinity and Immune Escape Hotspots. Int. J. Mol. Sci. 2025, 26, 1507. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J. Phys. Chem. B 2021, 125, 850–873. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; Giron, C.C.; Laaksonen, A. Electrostatic Features for the Receptor Binding Domain of SARS-COV-2 Wildtype and Its Variants. Compass to the Severity of the Future Variants with the Charge-Rule. J. Phys. Chem. B 2022, 126, 6835–6852. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Kwon, P.S.; Oh, H.; Kwon, S.-J.; Jin, W.; Zhang, F.; Fraser, K.; Hong, J.J.; Linhardt, R.J.; Dordick, J.S. Sulfated Polysaccharides Effectively Inhibit SARS-CoV-2 in Vitro. Cell Discov. 2020, 6, 50. [Google Scholar] [CrossRef]
- Jian, F.; Wang, J.; Yisimayi, A.; Song, W.; Xu, Y.; Chen, X.; Niu, X.; Yang, S.; Yu, Y.; Wang, P.; et al. Evolving Antibody Response to SARS-CoV-2 Antigenic Shift from XBB to JN.1. Nature 2025, 637, 921–929. [Google Scholar] [CrossRef]
- Taylor, A.L.; Starr, T.N. Deep Mutational Scanning of SARS-CoV-2 Omicron BA.2.86 and Epistatic Emergence of the KP.3 Variant. Virus Evol. 2024, 10, veae067. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Sun, Z.; Zhang, Y.; Jian, F.; Yang, S.; Xia, K.; Yu, L.; Wang, J.; Shao, F.; Wang, X.; et al. Structural and Molecular Basis of the Epistasis Effect in Enhanced Affinity between SARS-CoV-2 KP.3 and ACE2. Cell Discov. 2024, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guo, H.; Wang, A.; Cao, L.; Fan, Q.; Jiang, J.; Wang, M.; Lin, L.; Ge, X.; Wang, H.; et al. Structural Basis for the Evolution and Antibody Evasion of SARS-CoV-2 BA.2.86 and JN.1 Subvariants. Nat. Commun. 2024, 15, 7715. [Google Scholar] [CrossRef]
- Yajima, H.; Nomai, T.; Okumura, K.; Maenaka, K.; Ito, J.; Hashiguchi, T.; Sato, K.; Matsuno, K.; Nao, N.; Sawa, H.; et al. Molecular and Structural Insights into SARS-CoV-2 Evolution: From BA.2 to XBB Subvariants. mBio 2024, 15, e0322023. [Google Scholar] [CrossRef]
- Xue, S.; Han, Y.; Wu, F.; Wang, Q. Mutations in the SARS-CoV-2 Spike Receptor Binding Domain and Their Delicate Balance between ACE2 Affinity and Antibody Evasion. Protein Cell. 2024, 15, 403–418. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A template based loop structure prediction server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Cheng, J. 3Drefine: Consistent Protein Structure Refinement by Optimizing Hydrogen Bonding Network and Atomic-Level Energy Minimization. Proteins 2013, 81, 119–131. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An Interactive Web Server for Efficient Protein Structure Refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. VMD Store-A VMD Plugin to Browse, Discover, and Install VMD Extensions. J. Chem. Inf. Model. 2019, 59, 4519–4523. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef]
- Dehouck, Y.; Gilis, D.; Rooman, M. A new generation of statistical potentials for proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks:PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A Web Server to Predict and Analyze the Protein–Protein Complex Based on Computational Docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Mongan, J.; Simmerling, C.; McCammon, J.A.; Case, D.A.; Onufriev, A. Generalized Born Model with a Simple, Robust Molecular Volume Correction. J. Chem. Theory Comput. 2007, 3, 156–169. [Google Scholar] [CrossRef]
- Williams, A.H.; Zhan, C.-G. Generalized Methodology for the Quick Prediction of Variant SARS-CoV-2 Spike Protein Binding Affinities with Human Angiotensin-Converting Enzyme II. J. Phys. Chem. B 2022, 126, 2353–2360. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 7. Entropy Effects on the Performance of End-Point Binding Free Energy Calculation Approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Koukos, P.I.; Glykos, N.M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I. Adaptability of protein structures to enable functional interactions and evolutionary implications. Curr. Opin. Struct. Biol. 2015, 35, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Doruker, P.; Kaynak, B.; Zhang, S.; Krieger, J.; Li, H.; Bahar, I. Intrinsic dynamics is evolutionarily optimized to enable allosteric behavior. Curr. Opin. Struct. Biol. 2020, 62, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Faraone, J.N.; Evans, J.P.; Zheng, Y.-M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Panchal, A.R.; et al. Enhanced Evasion of Neutralizing Antibody Response by Omicron XBB.1.5, CH.1.1, and CA.3.1 Variants. Cell Rep. 2023, 42, 112443. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Faraone, J.N.; Hsu, C.C.; Chamblee, M.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; Saif, L.J.; et al. Neutralization Escape, Infectivity, and Membrane Fusion of JN.1-Derived SARS-CoV-2 SLip, FLiRT, and KP.2 Variants. Cell Rep. 2024, 43, 114520. [Google Scholar] [CrossRef]
- Li, P.; Liu, Y.; Faraone, J.N.; Hsu, C.C.; Chamblee, M.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; et al. Distinct Patterns of SARS-CoV-2 BA.2.87.1 and JN.1 Variants in Immune Evasion, Antigenicity, and Cell-Cell Fusion. mBio 2024, 15, e0075124. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective Mapping of Viral Mutations That Escape Antibodies Used to Treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Sartor, I.T.S.; Varela, F.H.; Meireles, M.R.; Kern, L.B.; Azevedo, T.R.; Giannini, G.L.T.; da Silva, M.S.; Demoliner, M.; Gularte, J.S.; de Almeida, P.R.; et al. Y380Q Novel Mutation in Receptor-Binding Domain of SARS-CoV-2 Spike Protein Together with C379W Interfere in the Neutralizing Antibodies Interaction. Diagn. Microbiol. Infect. Dis. 2022, 102, 115636. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, M.; Dhama, K.; Lee, S.-S.; Chakraborty, C. Can the RBD Mutation R346X Provide an Additional Fitness to the “Variant Soup,” Including Offspring of BQ and XBB of SARS-CoV-2 Omicron for the Antibody Resistance? Mol. Ther. Nucleic Acids 2023, 32, 61–63. [Google Scholar] [CrossRef]
- Mohandas, S.; Yadav, P.D.; Sapkal, G.; Shete, A.M.; Deshpande, G.; Nyayanit, D.A.; Patil, D.; Kadam, M.; Kumar, A.; Mote, C.; et al. Pathogenicity of SARS-CoV-2 Omicron (R346K) Variant in Syrian Hamsters and Its Cross-Neutralization with Different Variants of Concern. eBioMedicine 2022, 79, 103997. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshahrani, M.; Parikh, V.; Foley, B.; Verkhivker, G. Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape. Viruses 2025, 17, 741. https://doi.org/10.3390/v17060741
Alshahrani M, Parikh V, Foley B, Verkhivker G. Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape. Viruses. 2025; 17(6):741. https://doi.org/10.3390/v17060741
Chicago/Turabian StyleAlshahrani, Mohammed, Vedant Parikh, Brandon Foley, and Gennady Verkhivker. 2025. "Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape" Viruses 17, no. 6: 741. https://doi.org/10.3390/v17060741
APA StyleAlshahrani, M., Parikh, V., Foley, B., & Verkhivker, G. (2025). Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape. Viruses, 17(6), 741. https://doi.org/10.3390/v17060741