
Recommendations for Uniform Variant Calling of SARS-CoV-2 Genome Sequence across Bioinformatic Workflows

 , , , , , , , , , and
, , , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Datasets
2.2. SARS-CoV-2 Bioinformatic Workflow Overview
2.3. Data Pre-Processing Impacts on Variant Calling
2.4. A Parsimony Normalization Method to Standardize Variant Reporting across All Workflows Allowed Vis-a-Vis Comparison of Variants
2.5. Within-Sample Allele Frequency and Per-Position Depth of Coverage Determine the Consistency of Variant Calls across Workflows
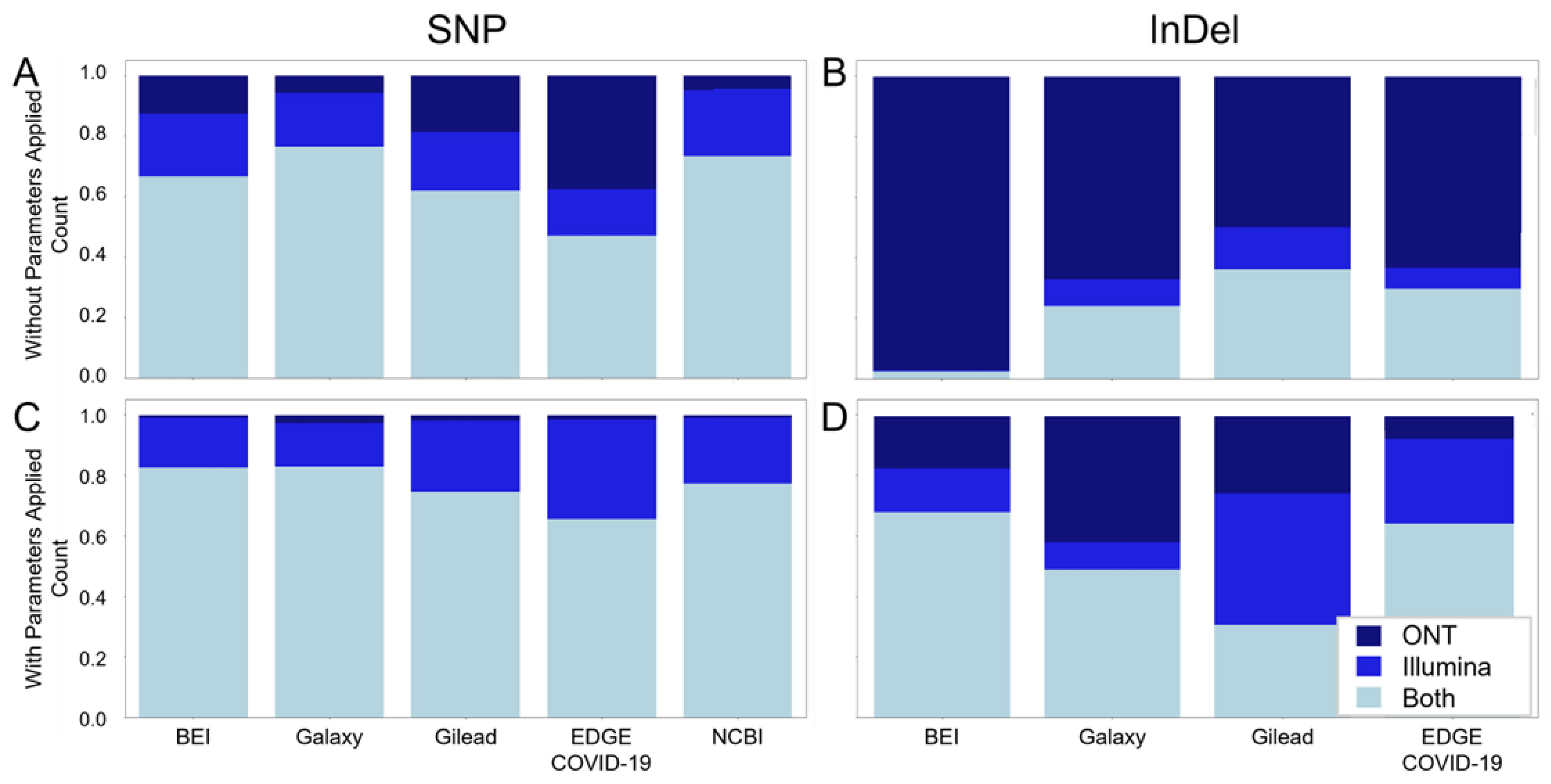
2.6. Filtering for Highly Supported Variant Calls Supports Cross-Workflow and Cross-Platform Agreement
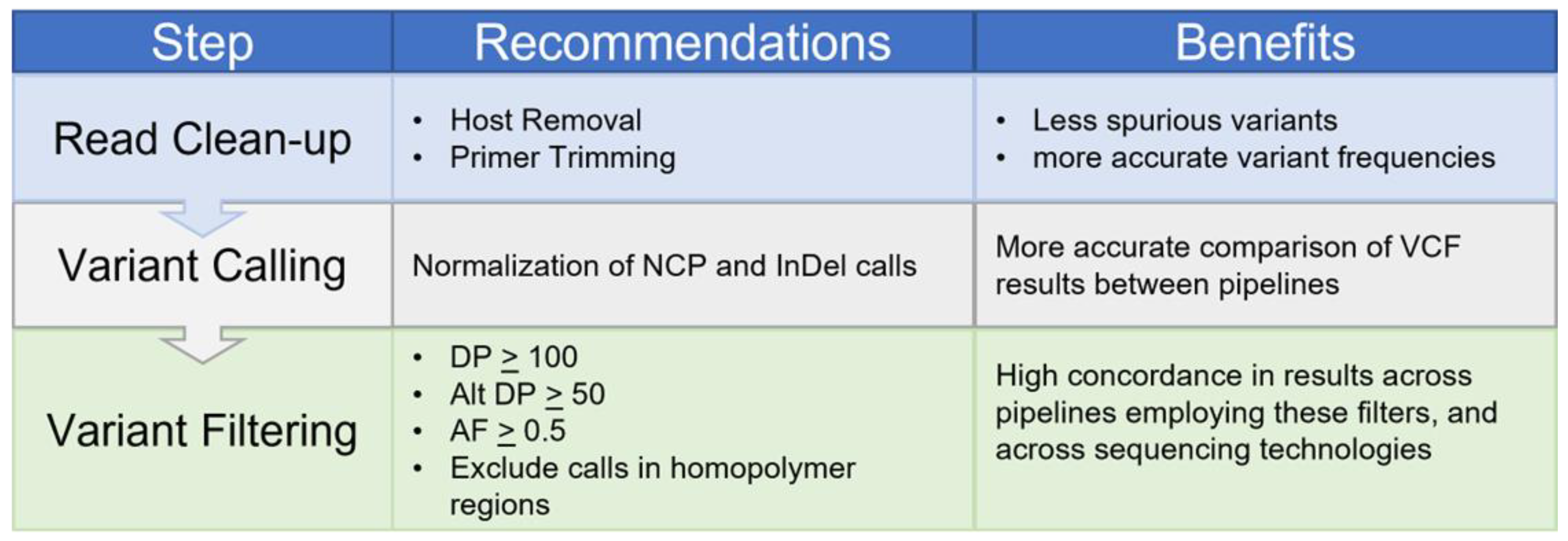
3. Discussion
4. Methods
4.1. Dataset
4.2. BEI Resources
4.3. Galaxy Project
4.3.1. Variant Calling from Paired-End Illumina Short-Read Data
4.3.2. Variant Calling from Oxford Nanopore Long-Read Data
4.4. Gilead Sciences
4.4.1. Illumina
4.4.2. ONT
4.5. Los Alamos National Laboratory (EDGE-COVID19)
4.6. Lilly
Illumina
4.7. NCBI
4.7.1. Illumina
4.7.2. ONT
4.8. VIR
4.8.1. Illumina
4.8.2. ONT
5. Parsimony Script
6. Studying the Effects of Primer Trimming on Variant Calls and Apparent Allele Frequencies of Called Variants
7. Calculation of Receiver Operating Characteristic (ROC) Plots Based on Concordance across Workflows
8. Calculation of Receiver Operating Characteristic (ROC) Plots Based on Cross-Platform Agreement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khoury, M.J.; Bowen, M.S.; Clyne, M.; Dotson, W.D.; Gwinn, M.L.; Green, R.F.; Kolor, K.; Rodriguez, J.L.; Wulf, A.; Yu, W. From Public Health Genomics to Precision Public Health: A 20-Year Journey. Genet. Med. 2018, 20, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, N.; Descamps, T.; Devleesschauwer, B.; Roosens, N.H.C.; Boon, N.A.M.; Van Oyen, H.; Robert, A. Status and Potential of Bacterial Genomics for Public Health Practice: A Scoping Review. Implement. Sci. 2019, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. The Sequence Read Archive: A Decade More of Explosive Growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.W.; Jamrozy, D. Genomics and Epidemiological Surveillance. Nat. Rev. Microbiol. 2020, 18, 478. [Google Scholar] [CrossRef]
- Ling-Hu, T.; Rios-Guzman, E.; Lorenzo-Redondo, R.; Ozer, E.A.; Hultquist, J.F. Challenges and Opportunities for Global Genomic Surveillance Strategies in the COVID-19 Era. Viruses 2022, 14, 2532. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next Generation Sequencing of SARS-CoV-2 Genomes: Challenges, Applications and Opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Zverinova, S.; Guryev, V. Variant Calling: Considerations, Practices, and Developments. Hum. Mutat. 2022, 43, 976–985. [Google Scholar] [CrossRef]
- Pan, B.; Ren, L.; Onuchic, V.; Guan, M.; Kusko, R.; Bruinsma, S.; Trigg, L.; Scherer, A.; Ning, B.; Zhang, C.; et al. Assessing Reproducibility of Inherited Variants Detected with Short-Read Whole Genome Sequencing. Genome Biol. 2022, 23, 2. [Google Scholar] [CrossRef]
- Foox, J.; Tighe, S.W.; Nicolet, C.M.; Zook, J.M.; Byrska-Bishop, M.; Clarke, W.E.; Khayat, M.M.; Mahmoud, M.; Laaguiby, P.K.; Herbert, Z.T.; et al. Performance Assessment of DNA Sequencing Platforms in the ABRF Next-Generation Sequencing Study. Nat. Biotechnol. 2021, 39, 1129–1140. [Google Scholar] [CrossRef]
- Krishnan, V.; Utiramerur, S.; Ng, Z.; Datta, S.; Snyder, M.P.; Ashley, E.A. Benchmarking Workflows to Assess Performance and Suitability of Germline Variant Calling Pipelines in Clinical Diagnostic Assays. BMC Bioinform. 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Lincoln, S.E.; Truty, R.; Lin, C.F.; Zook, J.M.; Paul, J.; Ramey, V.H.; Salit, M.; Rehm, H.L.; Nussbaum, R.L.; Lebo, M.S. A Rigorous Interlaboratory Examination of the Need to Confirm Next-Generation Sequencing–Detected Variants with an Orthogonal Method in Clinical Genetic Testing. J. Mol. Diagn. 2019, 21, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Cornish, A.; Guda, C. A Comparison of Variant Calling Pipelines Using Genome in a Bottle as a Reference. Biomed Res. Int. 2015, 2015, 456479. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Chang, P.C.; Hsu, C.; Hung, M.Z.; Chien, Y.H.; Hwu, W.L.; Lai, F.P.; Lee, N.C. Comparison of GATK and DeepVariant by Trio Sequencing. Sci. Rep. 2022, 12, 1809. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Zhong, H.; Meng, Y.; Du, H. Systematic Comparison of Germline Variant Calling Pipelines Cross Multiple Next-Generation Sequencers. Sci. Rep. 2019, 9, 9345. [Google Scholar] [CrossRef] [PubMed]
- Krusche, P.; Trigg, L.; Boutros, P.C.; Mason, C.E.; La Vega, F.M.D.; Moore, B.L.; Gonzalez-Porta, M.; Eberle, M.A.; Tezak, Z.; Lababidi, S.; et al. Best Practices for Benchmarking Germline Small-Variant Calls in Human Genomes. Nat. Biotechnol. 2019, 37, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Ren, L.; Chen, Z.; Fang, L.T.; Zhao, Y.; Lack, J.; Guan, M.; Zhu, B.; Jaeger, E.; Kerrigan, L.; et al. Toward Best Practice in Cancer Mutation Detection with Whole-Genome and Whole-Exome Sequencing. Nat. Biotechnol. 2021, 39, 1141–1150. [Google Scholar] [CrossRef]
- Lo, C.-C.; Shakya, M.; Connor, R.; Davenport, K.; Flynn, M.; y Gutiérrez, A.M.; Hu, B.; Li, P.-E.; Jackson, E.P.; Xu, Y.; et al. EDGE COVID-19: A Web Platform to Generate Submission-Ready Genomes from SARS-CoV-2 Sequencing Efforts. Bioinformatics 2022, 38, 2700–2704. [Google Scholar] [CrossRef]
- Tracking Resistance and Coronavirus Evolution (TRACE)|National Institutes of Health (NIH). Available online: https://www.nih.gov/research-training/medical-research-initiatives/activ/tracking-resistance-coronavirus-evolution-trace (accessed on 25 August 2023).
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.S.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. STAT: A Fast, Scalable, MinHash-Based k-Mer Tool to Assess Sequence Read Archive next-Generation Sequence Submissions. Genome Biol. 2021, 22, 270. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.B.; Moyer, E.; Phan, L.; Maglott, D.; Kattman, B. SPDI: Data Model for Variants and Applications at NCBI. Bioinformatics 2020, 36, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Van Poelvoorde, L.A.E.; Delcourt, T.; Coucke, W.; Herman, P.; De Keersmaecker, S.C.J.; Saelens, X.; Roosens, N.H.C.; Vanneste, K. Strategy and Performance Evaluation of Low-Frequency Variant Calling for SARS-CoV-2 Using Targeted Deep Illumina Sequencing. Front. Microbiol. 2021, 12, 3073. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; Smith, K.; De Maio, N.; Haussler, D.; Lanfear, R.; et al. Pandemic-Scale Phylogenomics Reveals the SARS-CoV-2 Recombination Landscape. Nature 2022, 609, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, B.; Castelán Sánchez, H.G.; da Silva Candido, D.; Jackson, B.; Fleishon, S.; Houzet, R.; Ruis, C.; Delaye, L.; Faria, N.R.; Rambaut, A.; et al. Emergence and Widespread Circulation of a Recombinant SARS-CoV-2 Lineage in North America. Cell Host Microbe 2022, 30, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.J.; Draper, J.; Gall, M.; Sim, E.M.; Arnott, A.; Agius, J.E.; Johnson-Mackinnon, J.; Fong, W.; Martinez, E.; Drew, A.P.; et al. Co-Infection with SARS-CoV-2 Omicron and Delta Variants Revealed by Genomic Surveillance. Nat. Commun. 2022, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Quinn, J.; Pinsky, B.; Shah, N.H.; Brown, I. Rates of Co-Infection Between SARS-CoV-2 and Other Respiratory Pathogens. JAMA 2020, 323, 2085–2086. [Google Scholar] [CrossRef]
- Mohammed, K.S.; Kibinge, N.; Prins, P.; Agoti, C.N.; Cotten, M.; Nokes, D.J.; Brand, S.; Githinji, G. Evaluating the Performance of Tools Used to Call Minority Variants from Whole Genome Short-Read Data. Wellcome Open Res. 2018, 3. [Google Scholar] [CrossRef]
- Van Poelvoorde, L.A.E.; Delcourt, T.; Vuylsteke, M.; De Keersmaecker, S.C.J.; Thomas, I.; Van Gucht, S.; Saelens, X.; Roosens, N.; Vanneste, K. A General Approach to Identify Low-Frequency Variants within Influenza Samples Collected during Routine Surveillance. Microb. Genom. 2022, 8, mgen000867. [Google Scholar] [CrossRef]
- McCrone, J.T.; Lauring, A.S. Measurements of Intrahost Viral Diversity Are Extremely Sensitive to Systematic Errors in Variant Calling. J. Virol. 2016, 90, 6884. [Google Scholar] [CrossRef] [PubMed]
- Part 11, Electronic Records; Electronic Signatures—Scope and Application|FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application (accessed on 6 June 2023).
- Donaldson, E.F.; Deming, D.J.; O’Rear, J.J.; Naeger, L.K. Regulatory Evaluation of Antiviral Drug Resistance in the Era of Next-Generation Sequencing. Biomark. Med. 2015, 9, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Broeckhoven, C. Van NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Van der Auwera, G.; O’Connor, B. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Newton, MA, USA, 2020; ISBN 9781491975190. [Google Scholar]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- GitHub—Nanoporetech/Medaka: Sequence Correction Provided by ONT Research. Available online: https://github.com/nanoporetech/medaka (accessed on 3 October 2023).
- Afgan, E.; Nekrutenko, A.; Grüning, B.A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; Ostrovsky, A.E.; Mahmoud, A.; Lonie, A.J.; Syme, A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Maier, W.; Bray, S.; van den Beek, M.; Bouvier, D.; Coraor, N.; Miladi, M.; Singh, B.; De Argila, J.R.; Baker, D.; Roach, N.; et al. Ready-to-Use Public Infrastructure for Global SARS-CoV-2 Monitoring. Nat. Biotechnol. 2021, 39, 1178–1179. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.D.; Yuen, D.; Chung, V.; Duncan, A.G.; Liu, X.K.; Patricia, J.; Paten, B.; Stein, L.; Ferretti, V. The Dockstore: Enabling Modular, Community-Focused Sharing of Docker-Based Genomics Tools and Workflows. F1000Res 2017, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Bray, S.; van den Beek, M.; Bouvier, D.; Coraor, N.; Miladi, M.; Singh, B.; De Argila, J.R.; Baker, D.; Roach, N.; et al. Freely Accessible Ready to Use Global Infrastructure for SARS-CoV-2 Monitoring. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Nickloman; Rowe, W.; Poplawski, R.; Rudd, S.; Rambaut, A.; Bedford, T.; Karthik, W.S.; Simpson, J. Artic-Network/Fieldbioinformatics: 1.2.2 2022. Available online: https://zenodo.org/records/6245266 (accessed on 4 December 2023).
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Gaspar, J.M. NGmerge: Merging Paired-End Reads via Novel Empirically-Derived Models of Sequencing Errors. BMC Bioinform. 2018, 19, 536. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Stancu, M.C.; Van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; De Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Valle-Inclan, J.E.; et al. Mapping and Phasing of Structural Variation in Patient Genomes Using Nanopore Sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From Squiggle to Basepair: Computational Approaches for Improving Nanopore Sequencing Read Accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156. [Google Scholar] [CrossRef]
- Li, H.; Barrett, J. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Connor, R.; Shakya, M.; Yarmosh, D.A.; Maier, W.; Martin, R.; Bradford, R.; Brister, J.R.; Chain, P.S.G.; Copeland, C.A.; di Iulio, J.; et al. Recommendations for Uniform Variant Calling of SARS-CoV-2 Genome Sequence across Bioinformatic Workflows. Viruses 2024, 16, 430. https://doi.org/10.3390/v16030430
Connor R, Shakya M, Yarmosh DA, Maier W, Martin R, Bradford R, Brister JR, Chain PSG, Copeland CA, di Iulio J, et al. Recommendations for Uniform Variant Calling of SARS-CoV-2 Genome Sequence across Bioinformatic Workflows. Viruses. 2024; 16(3):430. https://doi.org/10.3390/v16030430
Chicago/Turabian StyleConnor, Ryan, Migun Shakya, David A. Yarmosh, Wolfgang Maier, Ross Martin, Rebecca Bradford, J. Rodney Brister, Patrick S. G. Chain, Courtney A. Copeland, Julia di Iulio, and et al. 2024. "Recommendations for Uniform Variant Calling of SARS-CoV-2 Genome Sequence across Bioinformatic Workflows" Viruses 16, no. 3: 430. https://doi.org/10.3390/v16030430
APA StyleConnor, R., Shakya, M., Yarmosh, D. A., Maier, W., Martin, R., Bradford, R., Brister, J. R., Chain, P. S. G., Copeland, C. A., di Iulio, J., Hu, B., Ebert, P., Gunti, J., Jin, Y., Katz, K. S., Kochergin, A., LaRosa, T., Li, J., Li, P.-E., ... Pruitt, K. D. (2024). Recommendations for Uniform Variant Calling of SARS-CoV-2 Genome Sequence across Bioinformatic Workflows. Viruses, 16(3), 430. https://doi.org/10.3390/v16030430