

Molecular Mechanisms of Cell-to-Cell Transmission in Human Herpesviruses

Abstract
1. Introduction
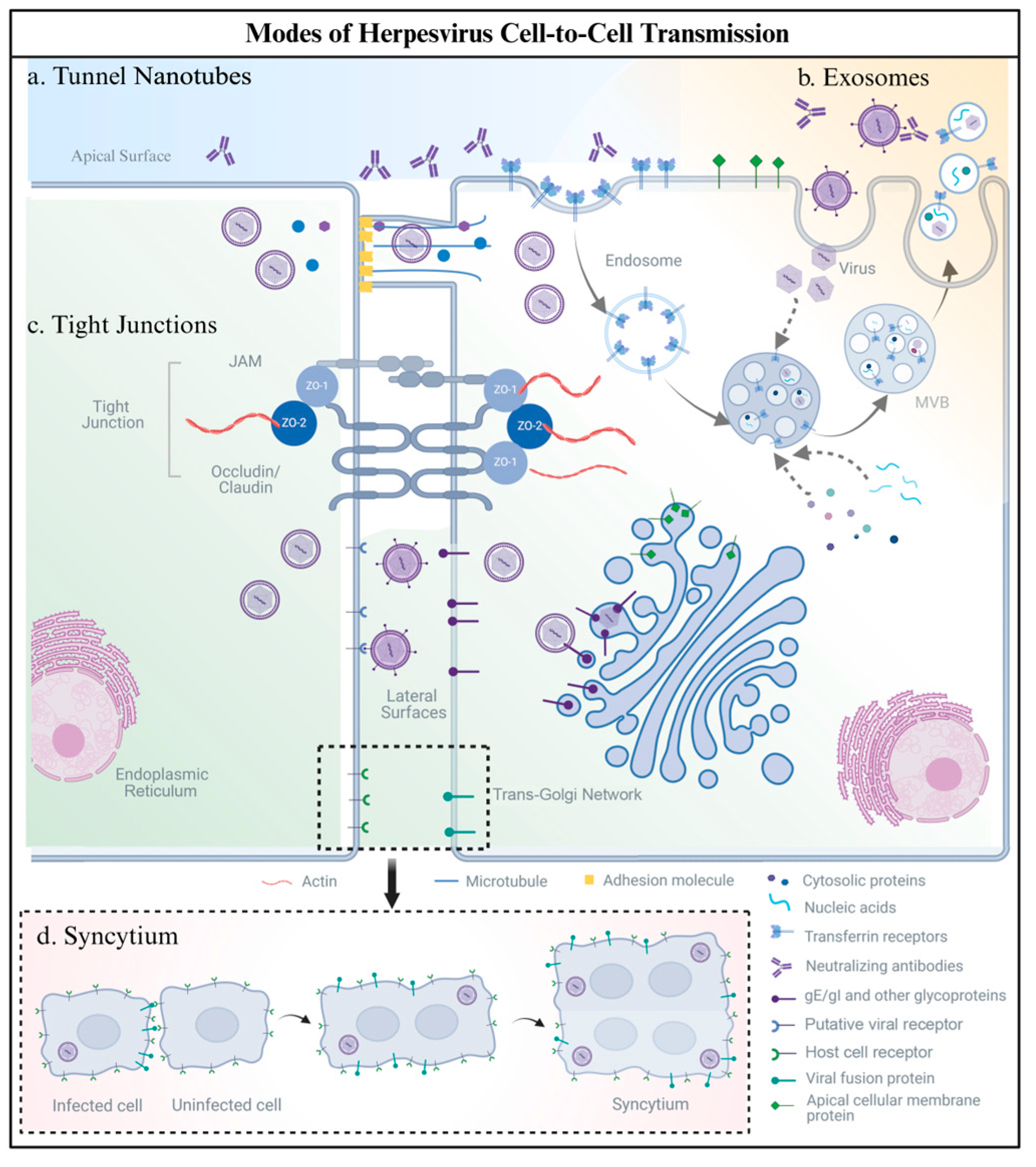
2. Modes of Human Herpesvirus Cell-to-Cell Transmission
2.1. Tunnel Nanotubes
2.2. Exosomes
2.3. Tight Junctions
2.4. Syncytium
3. Biological Advantages of Herpesvirus Cell-to-Cell Transmission
3.1. Facilitating Viral Latency and Reactivation
3.2. Enhanced Infection Efficiency and Immune Evasion
3.3. Augmented Pathogenicity and Tissue Invasion
4. Intervention Strategies Targeting Cell-to-Cell Transmission
4.1. Monoclonal Antibodies
4.2. Antiviral Compounds
4.3. Vaccines
5. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Wijesinghe, V.N.; Farouk, I.A.; Zabidi, N.Z.; Puniyamurti, A.; Choo, W.S.; Lal, S.K. Current vaccine approaches and emerging strategies against herpes simplex virus (HSV). Expert Rev. Vaccines 2021, 20, 1077–1096. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Yi, R.P.; Xu, S.T. Progress of molecular epidemiological research on human herpes virus. Int. J. Virol. 2021, 28, 170–173. [Google Scholar] [CrossRef]
- Jansens, R.J.J.; Tishchenko, A.; Favoreel, H.W. Bridging the Gap: Virus Long-Distance Spread via Tunneling Nanotubes. J. Virol. 2020, 94, e02120-19. [Google Scholar] [CrossRef] [PubMed]
- Jansens, R.J.J.; Van den Broeck, W.; De Pelsmaeker, S.; Lamote, J.A.S.; Van Waesberghe, C.; Couck, L.; Favoreel, H.W. Pseudorabies Virus US3-Induced Tunneling Nanotubes Contain Stabilized Microtubules, Interact with Neighboring Cells via Cadherins, and Allow Intercellular Molecular Communication. J. Virol. 2017, 91, e00749-17. [Google Scholar] [CrossRef]
- Ladelfa, M.F.; Kotsias, F.; Del Médico Zajac, M.P.; Van den Broeke, C.; Favoreel, H.; Romera, S.A.; Calamante, G. Effect of the US3 protein of bovine herpesvirus 5 on the actin cytoskeleton and apoptosis. Vet. Microbiol. 2011, 153, 361–366. [Google Scholar] [CrossRef]
- Panasiuk, M.; Rychłowski, M.; Derewońko, N.; Bieńkowska-Szewczyk, K. Tunneling Nanotubes as a Novel Route of Cell-to-Cell Spread of Herpesviruses. J. Virol. 2018, 92, e00090-18. [Google Scholar] [CrossRef]
- Wang, J.; Shang, K.T.; Ma, Q.H.; Dong, Z.Y.; Chen, Y.H.; Yao, Y.F. Herpes Simplex Virus Type 1 Infection Induces the Formation of Tunneling Nanotubes. Microorganisms 2023, 11, 1916. [Google Scholar] [CrossRef]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Hou, L.; Tang, Y.D.; Liu, Y.; Wang, S.; Wang, J.; Shen, N.; An, T.; Tian, Z.; Cai, X. Pseudorabies virus infection inhibits autophagy in permissive cells in vitro. Sci. Rep. 2017, 7, 39964. [Google Scholar] [CrossRef]
- Watanabe, M.; Arii, J.; Takeshima, K.; Fukui, A.; Shimojima, M.; Kozuka-Hata, H.; Oyama, M.; Minamitani, T.; Yasui, T.; Kubota, Y.; et al. Prohibitin-1 Contributes to Cell-to-Cell Transmission of Herpes Simplex Virus 1 via the MAPK/ERK Signaling Pathway. J. Virol. 2021, 95, e01413-20. [Google Scholar] [CrossRef]
- Huang, L.; Zhao, T.; Zhao, W.; Shao, A.; Zhao, H.; Ma, W.; Gong, Y.; Zeng, X.; Weng, C.; Bu, L.; et al. Herpes zoster mRNA vaccine induces superior vaccine immunity over licensed vaccine in mice and rhesus macaques. Emerg. Microbes Infect. 2024, 13, 2309985. [Google Scholar] [CrossRef] [PubMed]
- Lecrenier, N.; Beukelaers, P.; Colindres, R.; Curran, D.; De Kesel, C.; De Saegher, J.P.; Didierlaurent, A.M.; Ledent, E.Y.; Mols, J.F.; Mrkvan, T.; et al. Development of adjuvanted recombinant zoster vaccine and its implications for shingles prevention. Expert Rev. Vaccines 2018, 17, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef]
- Loesing, J.-B.; Di Fiore, S.; Ritter, K.; Fischer, R.; Kleines, M. Epstein–Barr virus BDLF2–BMRF2 complex affects cellular morphology. J. Gen. Virol. 2009, 90, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.B.; Edgar, R.; May, J.S.; Stevenson, P.G. A gamma-herpesvirus glycoprotein complex manipulates actin to promote viral spread. PLoS ONE 2008, 3, e1808. [Google Scholar] [CrossRef]
- Möhl, B.S.; Chen, J.; Park, S.J.; Jardetzky, T.S.; Longnecker, R. Epstein-Barr Virus Fusion with Epithelial Cells Triggered by gB Is Restricted by a gL Glycosylation Site. J. Virol. 2017, 91, e01255-17. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Deschamps, T. Extracellular vesicles during Herpes Simplex Virus type 1 infection: An inquire. Virol. J. 2016, 13, 63. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Extracellular Vesicles Released by Herpes Simplex Virus 1-Infected Cells Block Virus Replication in Recipient Cells in a STING-Dependent Manner. J. Virol. 2018, 92, e01102-18. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Dong, H.; Yang, Z.; Zhou, L.; Xu, P. PML Body Component Sp100A Restricts Wild-Type Herpes Simplex Virus 1 Infection. J. Virol. 2022, 96, e0027922. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, Z.; Li, Y.; Yang, M.; Hu, Q. Migrasomes released by HSV-2-infected cells serve as a conveyance for virus spread. Virol. Sin. 2023, 38, 643–645. [Google Scholar] [CrossRef]
- Streck, N.T.; Zhao, Y.; Sundstrom, J.M.; Buchkovich, N.J. Human Cytomegalovirus Utilizes Extracellular Vesicles To Enhance Virus Spread. J. Virol. 2020, 94, e00609-20. [Google Scholar] [CrossRef] [PubMed]
- Panther, L.; Basnet, S.; Fierro, C.; Brune, D.; Leggett, R.; Peterson, J.; Pickrell, P.; Lin, J.; Wu, K.; Lee, H.; et al. 2892. Safety and Immunogenicity of mRNA-1647, an mRNA-Based Cytomegalovirus Vaccine in Healthy Adults: Results of a Phase 2, Randomized, Observer-Blind, Placebo-Controlled, Dose-Finding Trial. Open Forum Infect. Dis. 2023, 10, ofad500.2475. [Google Scholar] [CrossRef]
- Ahmed, W.; Tariq, S.; Khan, G. Tracking EBV-encoded RNAs (EBERs) from the nucleus to the excreted exosomes of B-lymphocytes. Sci. Rep. 2018, 8, 15438. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, S. Advances on the mechanism of Epstein-Barr virus transmission in vivo. J. Clin. Pathol. Res. 2020, 40, 2727–2733. [Google Scholar] [CrossRef]
- McMillan, T.N.; Johnson, D.C. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 2001, 75, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Huber, M.T. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef]
- Farnsworth, A.; Johnson, D.C. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J. Virol. 2006, 80, 3167–3179. [Google Scholar] [CrossRef]
- Roller, R.J.; Haugo, A.C.; Yang, K.; Baines, J.D. The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J. Virol. 2014, 88, 4058–4068. [Google Scholar] [CrossRef]
- Roller, R.J.; Fetters, R. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J. Virol. 2015, 89, 3112–3122. [Google Scholar] [CrossRef]
- Feutz, E.; McLeland-Wieser, H.; Ma, J.; Roller, R.J. Functional interactions between herpes simplex virus pUL51, pUL7 and gE reveal cell-specific mechanisms for epithelial cell-to-cell spread. Virology 2019, 537, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, J.; Maringer, K.; Cook, R.; Bernard, E.; Elliott, G. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. J. Virol. 2009, 83, 5204–5218. [Google Scholar] [CrossRef] [PubMed]
- Maringer, K.; Stylianou, J.; Elliott, G. A network of protein interactions around the herpes simplex virus tegument protein VP22. J. Virol. 2012, 86, 12971–12982. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, A.; Wisner, T.W.; Johnson, D.C. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 2007, 81, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Bzik, D.J.; Fox, B.A.; DeLuca, N.A.; Person, S. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: Mutations affecting rate of virus entry and cell fusion. Virology 1984, 137, 185–190. [Google Scholar] [CrossRef]
- Delboy, M.G.; Patterson, J.L.; Hollander, A.M.; Nicola, A.V. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol. J. 2006, 3, 105. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, X.Q.; Zhang, B.; Gu, J.; Meng, F.Z.; Liu, H.; Zhou, L.; Wang, X.; Hou, W.; Ho, W.Z. Bowman-Birk Inhibitor Suppresses Herpes Simplex Virus Type 2 Infection of Human Cervical Epithelial Cells. Viruses 2018, 10, 557. [Google Scholar] [CrossRef]
- Xu, X.Q.; Liu, Y.; Zhang, B.; Liu, H.; Shao, D.D.; Liu, J.B.; Wang, X.; Zhou, L.N.; Hu, W.H.; Ho, W.Z. IL-22 suppresses HSV-2 replication in human cervical epithelial cells. Cytokine 2019, 123, 154776. [Google Scholar] [CrossRef]
- Kalu, N.N.; Desai, P.J.; Shirley, C.M.; Gibson, W.; Dennis, P.A.; Ambinder, R.F. Nelfinavir inhibits maturation and export of herpes simplex virus 1. J. Virol. 2014, 88, 5455–5461. [Google Scholar] [CrossRef]
- Su, C.; Wu, L.; Chai, Y.; Qi, J.; Tan, S.; Gao, G.F.; Song, H.; Yan, J. Molecular basis of EphA2 recognition by gHgL from gammaherpesviruses. Nat. Commun. 2020, 11, 5964. [Google Scholar] [CrossRef]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of Events Governing Cell-Cell Fusion Induced by Herpes Simplex Virus Glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar] [CrossRef] [PubMed]
- Terry-Allison, T.; Montgomery, R.I.; Whitbeck, J.C.; Xu, R.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. HveA (Herpesvirus Entry Mediator A), a Coreceptor for Herpes Simplex Virus Entry, also Participates in Virus-Induced Cell Fusion. J. Virol. 1998, 72, 5802–5810. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.C.; Wills, J.W. Differential Requirements for gE, gI, and UL16 among Herpes Simplex Virus 1 Syncytial Variants Suggest Unique Modes of Dysregulating the Mechanism of Cell-to-Cell Spread. J. Virol. 2019, 93, e00494-19. [Google Scholar] [CrossRef]
- Maresova, L.; Pasieka, T.J.; Grose, C. Varicella-zoster Virus gB and gE coexpression, but not gB or gE alone, leads to abundant fusion and syncytium formation equivalent to those from gH and gL coexpression. J. Virol. 2001, 75, 9483–9492. [Google Scholar] [CrossRef]
- Liu, Y.; Guan, X.; Li, C.; Ni, F.; Luo, S.; Wang, J.; Zhang, D.; Zhang, M.; Hu, Q. HSV-2 glycoprotein J promotes viral protein expression and virus spread. Virology 2018, 525, 83–95. [Google Scholar] [CrossRef]
- Ruan, P.; Wang, M.; Cheng, A.; Zhao, X.; Yang, Q.; Wu, Y.; Zhang, S.; Tian, B.; Huang, J.; Ou, X.; et al. Mechanism of herpesvirus UL24 protein regulating viral immune escape and virulence. Front. Microbiol. 2023, 14, 1268429. [Google Scholar] [CrossRef]
- Carmichael, J.C.; Yokota, H.; Craven, R.C.; Schmitt, A.; Wills, J.W. The HSV-1 mechanisms of cell-to-cell spread and fusion are critically dependent on host PTP1B. PLoS Pathog. 2018, 14, e1007054. [Google Scholar] [CrossRef]
- Karasneh, G.A.; Ali, M.; Shukla, D. An important role for syndecan-1 in herpes simplex virus type-1 induced cell-to-cell fusion and virus spread. PLoS ONE 2011, 6, e25252. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Wang, L.; Xu, H.; Wang, Z.; Zhang, T.; Wang, M.; Ning, Y.; Deng, F.; Hu, Z.; Wang, H.; et al. A novel glycoprotein D-specific monoclonal antibody neutralizes herpes simplex virus. Antivir. Res. 2017, 147, 131–141. [Google Scholar] [CrossRef]
- Tian, R.; Ju, F.; Yu, M.; Liang, Z.; Xu, Z.; Zhao, M.; Qin, Y.; Lin, Y.; Huang, X.; Chang, Y.; et al. A potent neutralizing and protective antibody against a conserved continuous epitope on HSV glycoprotein D. Antivir. Res. 2022, 201, 105298. [Google Scholar] [CrossRef]
- Seyfizadeh, N.; Kalbermatter, D.; Imhof, T.; Ries, M.; Müller, C.; Jenner, L.; Blumenschein, E.; Yendrzheyevskiy, A.; Grün, F.; Moog, K.; et al. Development of a highly effective combination monoclonal antibody therapy against Herpes simplex virus. J. Biomed. Sci. 2024, 31, 56. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, E.R.; Compton, T. Characterization of human cytomegalovirus glycoprotein-induced cell-cell fusion. J. Virol. 2005, 79, 7827–7837. [Google Scholar] [CrossRef]
- Cimato, G.; Zhou, X.; Brune, W.; Frascaroli, G. Human cytomegalovirus glycoprotein variants governing viral tropism and syncytium formation in epithelial cells and macrophages. J. Virol. 2024, 98, e0029324. [Google Scholar] [CrossRef]
- Zhang, X.; Hong, J.; Zhong, L.; Wu, Q.; Zhang, S.; Zhu, Q.; Chen, H.; Wei, D.; Li, R.; Zhang, W.; et al. Protective anti-gB neutralizing antibodies targeting two vulnerable sites for EBV-cell membrane fusion. Proc. Natl. Acad. Sci. USA 2022, 119, e2202371119. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Zurzolo, C. Tunneling nanotubes: Reshaping connectivity. Curr. Opin. Cell Biol. 2021, 71, 139–147. [Google Scholar] [CrossRef]
- Lv, W.; Li, Z.; Wang, S.; He, J.; Zhang, L. A role for tunneling nanotubes in virus spread. Front. Microbiol. 2024, 15, 1356415. [Google Scholar] [CrossRef]
- Wang, X.-Q.; Yu, X.-W.; Liu, M.-Y.; Guan, Y.-T. Progress in promoting mechanism of tunneling nanotubes formation. Acad. J. Second. Mil. Med. Univ. 2015, 36, 4. [Google Scholar] [CrossRef]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef]
- Cifuentes-Munoz, N.; El Najjar, F.; Dutch, R.E. Viral cell-to-cell spread: Conventional and non-conventional ways. Adv. Virus Res. 2020, 108, 85–125. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yamamoto, T.; Aoyama, Y.; Fujimoto, W. Cell-to-cell transmission of HSV-1 in differentiated keratinocytes promotes multinucleated giant cell formation. J. Dermatol. Sci. 2019, 93, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, Q.; Pan, D.; Yu, H.; Fu, W.; Liu, J.; He, M.; Zhu, R.; Cai, Y.; Huang, Y.; et al. Near-atomic cryo-electron microscopy structures of varicella-zoster virus capsids. Nat. Microbiol. 2020, 5, 1542–1552. [Google Scholar] [CrossRef]
- Yamamoto, T.; Aoyama, Y. Detection of multinucleated giant cells in differentiated keratinocytes with herpes simplex virus and varicella zoster virus infections by modified Tzanck smear method. J. Dermatol. 2020, 48, 21–27. [Google Scholar] [CrossRef]
- Tang, J.; Frascaroli, G.; Zhou, X.; Knickmann, J.; Brune, W. Cell Fusion and Syncytium Formation in Betaherpesvirus Infection. Viruses 2021, 13, 1973. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Bissinger, A.L.; Viebahn, R.; Oettle, H.; Radke, C.; Schmidt, C.A.; Jahn, G. Hepatocytes are permissive for human cytomegalovirus infection in human liver cell culture and In vivo. J. Infect. Dis. 1999, 180, 976–986. [Google Scholar] [CrossRef]
- Booth, J.C.; Beesley, J.E.; Stern, H. Syncytium formation caused by human cytomegalovirus in human embryonic lung fibroblasts. Arch. Virol. 1978, 57, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Percivalle, E.; Perez, L.; Lanzavecchia, A.; Lilleri, D. Monoclonal Antibodies to Different Components of the Human Cytomegalovirus (HCMV) Pentamer gH/gL/pUL128L and Trimer gH/gL/gO as well as Antibodies Elicited during Primary HCMV Infection Prevent Epithelial Cell Syncytium Formation. J. Virol. 2016, 90, 6216–6223. [Google Scholar] [CrossRef]
- Vo, M.; Aguiar, A.; McVoy, M.A.; Hertel, L. Cytomegalovirus Strain TB40/E Restrictions and Adaptations to Growth in ARPE-19 Epithelial Cells. Microorganisms 2020, 8, 615. [Google Scholar] [CrossRef]
- Bayliss, G.J.; Wolf, H. An Epstein--Barr virus early protein induces cell fusion. Proc. Natl. Acad. Sci. USA 1981, 78, 7162–7165. [Google Scholar] [CrossRef]
- Bello-Morales, R.; López-Guerrero, J.A. Extracellular Vesicles in Herpes Viral Spread and Immune Evasion. Front. Microbiol. 2018, 9, 2572. [Google Scholar] [CrossRef] [PubMed]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2021, 10, 617578. [Google Scholar] [CrossRef] [PubMed]
- Pegg, C.E.; Zaichick, S.V.; Bomba-Warczak, E.; Jovasevic, V.; Kim, D.; Kharkwal, H.; Wilson, D.W.; Walsh, D.; Sollars, P.J.; Pickard, G.E.; et al. Herpesviruses assimilate kinesin to produce motorized viral particles. Nature 2021, 599, 662–666. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.; Connor, V.; Coleman, H.M.; Proença, J.T.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Proença, J.T.; Connor, V.; Efstathiou, S. Influence of herpes simplex virus 1 latency-associated transcripts on the establishment and maintenance of latency in the ROSA26R reporter mouse model. J. Virol. 2012, 86, 8848–8858. [Google Scholar] [CrossRef]
- Polakovicova, I.; Jerez, S.; Wichmann, I.A.; Sandoval-Bórquez, A.; Carrasco-Véliz, N.; Corvalán, A.H. Role of microRNAs and Exosomes in Helicobacter pylori and Epstein-Barr Virus Associated Gastric Cancers. Front. Microbiol. 2018, 9, 636. [Google Scholar] [CrossRef]
- Wen, F.; Han, Y.; Zhang, H.; Zhao, Z.; Wang, W.; Chen, F.; Qin, W.; Ju, J.; An, L.; Meng, Y.; et al. Epstein-Barr virus infection upregulates extracellular OLFM4 to activate YAP signaling during gastric cancer progression. Nat. Commun. 2024, 15, 10543. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, A.E.; Enquist, L.W. Cell-Fusion Events Induced by α-Herpesviruses. Future Virol. 2015, 10, 185–200. [Google Scholar] [CrossRef]
- Sarfo, A.; Starkey, J.; Mellinger, E.; Zhang, D.; Chadha, P.; Carmichael, J.; Wills, J.W. The UL21 Tegument Protein of Herpes Simplex Virus 1 Is Differentially Required for the Syncytial Phenotype. J. Virol. 2017, 91, e01161-17. [Google Scholar] [CrossRef]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. USA 2005, 102, 8990–8995. [Google Scholar] [CrossRef]
- Temme, S.; Eis-Hübinger, A.M.; McLellan, A.D.; Koch, N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J. Immunol. 2010, 184, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Heilingloh, C.S.; Kummer, M.; Mühl-Zürbes, P.; Drassner, C.; Daniel, C.; Klewer, M.; Steinkasserer, A. L Particles Transmit Viral Proteins from Herpes Simplex Virus 1-Infected Mature Dendritic Cells to Uninfected Bystander Cells, Inducing CD83 Downmodulation. J. Virol. 2015, 89, 11046–11055. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Krawczyk, A. Role of L-Particles during Herpes Simplex Virus Infection. Front. Microbiol. 2017, 8, 2565. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, L.; Yang, B.; Li, D.; Wang, X.; Liu, X.; Tian, N.; Huang, Q.; Feng, R.; Wang, Z.; et al. Cutting Edge: Inhibition of Glycogen Synthase Kinase 3 Activity Induces the Generation and Enhanced Suppressive Function of Human IL-10(+) FOXP3(+)-Induced Regulatory T Cells. J. Immunol. 2020, 205, 1497–1502. [Google Scholar] [CrossRef]
- Bi, P.; Wang, J.; Lu, J.; Liu, X.; Wang, F.; Luo, Y.F.; Peng, S.D.; Li, X.P. Correlation analysis of the Epstein-Barr virus recruited regulatory T cells in nasopharyngeal carcinoma of immune escape. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2017, 52, 692–697. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Gunawardena, H.P.; Dekroon, R.M.; Heaton, P.R.; Edwards, R.H.; Ozgur, S.; Griffith, J.D.; Damania, B.; Raab-Traub, N. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc. Natl. Acad. Sci. USA 2013, 110, E2925–E2933. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C. Chapter Two—Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. In Advances in Virus Research; Kielian, M., Maramorosch, K., Mettenleiter, T.C., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 94, pp. 53–80. [Google Scholar]
- McCarthy, K.M.; Tank, D.W.; Enquist, L.W. Pseudorabies virus infection alters neuronal activity and connectivity in vitro. PLoS Pathog. 2009, 5, e1000640. [Google Scholar] [CrossRef]
- Laval, K.; Maturana, C.J.; Enquist, L.W. Mouse Footpad Inoculation Model to Study Viral-Induced Neuroinflammatory Responses. J. Vis. Exp. 2020, e61121. [Google Scholar] [CrossRef]
- Laval, K.; Van Cleemput, J.; Vernejoul, J.B.; Enquist, L.W. Alphaherpesvirus infection of mice primes PNS neurons to an inflammatory state regulated by TLR2 and type I IFN signaling. PLoS Pathog. 2019, 15, e1008087. [Google Scholar] [CrossRef]
- Wang, F.; Wu, J.-M.; Zhou, Y.-B.; Li, S.-Y.; Cheng, Y.-Z.; Qi, M.; Tang, W.; Wang, H.; Liu, J.; Yu, H. Mechanisms of vascular endothelial cell injury induced by human cytomegalovirus. J. Shandong Univ. (Health Sci.) 2007, 45, 1081–1084. [Google Scholar]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB signaling in inflammation and cancer. MedComm (2020) 2021, 2, 618–653. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Meng, Y.; LI, Y. Research advances in cytomegalovirus hepatitis. J. Clin. Hepatol. 2023, 39, 1431–1439. [Google Scholar] [CrossRef]
- Da Cunha, T.; Wu, G.Y. Cytomegalovirus Hepatitis in Immunocompetent and Immunocompromised Hosts. J. Clin. Transl. Hepatol. 2021, 9, 106–115. [Google Scholar] [CrossRef]
- Hong, J.; Zhong, L.; Liu, L.; Wu, Q.; Zhang, W.; Chen, K.; Wei, D.; Sun, H.; Zhou, X.; Zhang, X.; et al. Non-overlapping epitopes on the gHgL-gp42 complex for the rational design of a triple-antibody cocktail against EBV infection. Cell Rep. Med. 2023, 4, 101296. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.; Finlay, W.J.; Hearty, S.; O’Kennedy, R. Optimizing recombinant antibody function in SPR immunosensing. The influence of antibody structural format and chip surface chemistry on assay sensitivity. Biosens. Bioelectron. 2006, 22, 268–274. [Google Scholar] [CrossRef]
- Singh, A.; Pasha, S.K.; Manickam, P.; Bhansali, S. Single-domain antibody based thermally stable electrochemical immunosensor. Biosens. Bioelectron. 2016, 83, 162–168. [Google Scholar] [CrossRef]
- Monson, E.A.; Lloyd, M.G.; Johnson, R.I.; Caracciolo, K.; Whan, J.; Rau, T.F.; Londrigan, S.L.; Moffat, J.F.; Mayfosh, A.J.; Helbig, K.J. GS-1 blocks entry of herpes viruses and more broadly inhibits enveloped viruses. Antiviral Res. 2025, 237, 106136. [Google Scholar] [CrossRef]
- Cohen, J.I. The Varicella-Zoster Virus Genome. In Varicella-Zoster Virus; Abendroth, A., Arvin, A.M., Moffat, J.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–14. [Google Scholar]
- Duus, K.M.; Grose, C. Multiple regulatory effects of varicella-zoster virus (VZV) gL on trafficking patterns and fusogenic properties of VZV gH. J. Virol. 1996, 70, 8961–8971. [Google Scholar] [CrossRef]
- Zhao, G.X.; Bu, G.L.; Liu, G.F.; Kong, X.W.; Sun, C.; Li, Z.Q.; Dai, D.L.; Sun, H.X.; Kang, Y.F.; Feng, G.K.; et al. mRNA-based Vaccines Targeting the T-cell Epitope-rich Domain of Epstein Barr Virus Latent Proteins Elicit Robust Anti-Tumor Immunity in Mice. Adv. Sci. 2023, 10, e2302116. [Google Scholar] [CrossRef]
- Liu, H.; Chen, H.; Liu, Z.; Le, Z.; Nie, T.; Qiao, D.; Su, Y.; Mai, H.; Chen, Y.; Liu, L. Therapeutic nanovaccines sensitize EBV-associated tumors to checkpoint blockade therapy. Biomaterials 2020, 255, 120158. [Google Scholar] [CrossRef]
- Zhong, L.; Zhang, W.; Liu, H.; Zhang, X.; Yang, Z.; Wen, Z.; Chen, L.; Chen, H.; Luo, Y.; Chen, Y.; et al. A cocktail nanovaccine targeting key entry glycoproteins elicits high neutralizing antibody levels against EBV infection. Nat. Commun. 2024, 15, 5310. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Chin, A.L.; Liu, J.; Jardetzky, T.S.; Mudd, J.O.; Orloff, S.L.; Streblow, D.; Mussi-Pinhata, M.M.; Yamamoto, A.Y.; Duarte, G.; et al. HCMV trimer- and pentamer-specific antibodies synergize for virus neutralization but do not correlate with congenital transmission. Proc. Natl. Acad. Sci. USA 2019, 116, 3728–3733. [Google Scholar] [CrossRef]
- Xie, Z.; Song, Y. Research strategies and advances in vaccines against human cytomegalovirus infection. Prog. Microbiol. Immunol. 2023, 56–63. [Google Scholar]
- Hu, X.; Karthigeyan, K.P.; Herbek, S.; Valencia, S.M.; Jenks, J.A.; Webster, H.; Miller, I.G.; Connors, M.; Pollara, J.; Andy, C.; et al. Human Cytomegalovirus mRNA-1647 Vaccine Candidate Elicits Potent and Broad Neutralization and Higher Antibody-Dependent Cellular Cytotoxicity Responses Than the gB/MF59 Vaccine. J. Infect. Dis. 2024, 230, 455–466. [Google Scholar] [CrossRef]
- Johnston, C.; Gottlieb, S.L.; Wald, A. Status of vaccine research and development of vaccines for herpes simplex virus. Vaccine 2016, 34, 2948–2952. [Google Scholar] [CrossRef] [PubMed]
- Spicknall, I.H.; Looker, K.J.; Gottlieb, S.L.; Chesson, H.W.; Schiffer, J.T.; Elmes, J.; Boily, M.C. Review of mathematical models of HSV-2 vaccination: Implications for vaccine development. Vaccine 2019, 37, 7396–7407. [Google Scholar] [CrossRef]
- Stanberry, L.R.; Spruance, S.L.; Cunningham, A.L.; Bernstein, D.I.; Mindel, A.; Sacks, S.; Tyring, S.; Aoki, F.Y.; Slaoui, M.; Denis, M.; et al. Glycoprotein-D–Adjuvant Vaccine to Prevent Genital Herpes. N. Engl. J. Med. 2002, 347, 1652–1661. [Google Scholar] [CrossRef]
- Pražák, V.; Mironova, Y.; Vasishtan, D.; Hagen, C.; Laugks, U.; Jensen, Y.; Sanders, S.; Heumann, J.M.; Bosse, J.B.; Klupp, B.G.; et al. Molecular plasticity of herpesvirus nuclear egress analysed in situ. Nat. Microbiol. 2024, 9, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Wang, X.; Yang, S.; Liu, Y.; Zhang, G.; Zhuang, G. A rapid and accurate method for herpesviral gnome editing. Chin. J. Biotechnol. 2021, 37, 1376–1384. [Google Scholar] [CrossRef]
- Wu, H.; Li, T.; Zeng, M.; Peng, T. Herpes simplex virus type 1 infection activates the Epstein-Barr virus replicative cycle via a CREB-dependent mechanism. Cell Microbiol. 2012, 14, 546–559. [Google Scholar] [CrossRef]
- Botting, R.A.; Rana, H.; Bertram, K.M.; Rhodes, J.W.; Baharlou, H.; Nasr, N.; Cunningham, A.L.; Harman, A.N. Langerhans cells and sexual transmission of HIV and HSV. Rev. Med. Virol. 2017, 27, e1923. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.F.; Tobler, K.; Michaelsen, K.; Vogt, B.; Henckaerts, E.; Fraefel, C. Herpes Simplex Virus 1 Coinfection Modifies Adeno-associated Virus Genome End Recombination. J. Virol. 2021, 95, e0048621. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.F.; Tobler, K.; Leisi, R.; Lkharrazi, A.; Ros, C.; Fraefel, C. Herpes simplex virus co-infection facilitates rolling circle replication of the adeno-associated virus genome. PLoS Pathog. 2021, 17, e1009638. [Google Scholar] [CrossRef] [PubMed]
- Ahmadivand, S.; Soltani, M.; Shokrpoor, S.; Rahmati-Holasoo, H.; El-Matbouli, M.; Taheri-Mirghaed, A. Cyprinid herpesvirus 3 (CyHV-3) transmission and outbreaks in Iran: Detection and characterization in farmed common carp. Microb. Pathog. 2020, 149, 104321. [Google Scholar] [CrossRef]

{kind=link}
| Subfamily | Genus | Human Virus Species | Virus Name | Abbreviation |
|---|---|---|---|---|
| Alphaherpesvirinae | Simplexvirus | Simplexvirus humanalpha1 | human alphaherpesvirus 1 (Herpes simplex virus type 1) | HSV-1 |
| Simplexvirus humanalpha2 | human alphaherpesvirus 2 (Herpes simplex virus type 2) | HSV-2 | ||
| Varicellovirus | Varicellovirus humanalpha3 | human alphaherpesvirus 3 (varicella-zoster virus) | VZV | |
| Betaherpesvirinae | Cytomegalovirus | Cytomegalovirus humanbeta5 | human betaherpesvirus 5 (human cytomegalovirus) | HCMV |
| Roseolovirus | Roseolovirus humanbeta6a | human betaherpesvirus 6A (human herpesvirus 6A) | HHV6A | |
| Roseolovirus humanbeta6b | human betaherpesvirus 6B (human herpesvirus 6B) | HHV6B | ||
| Roseolovirus humanbeta7 | human betaherpesvirus7 (human herpesvirus 7) | HHV7 | ||
| Gammaherpesvirinae | Lymphocryptovirus | Lymphocryptovirus humangamma4 | human gammaherpesvirus 4 (Epstein–Barr virus) | EBV |
| Rhadinovirus | Rhadinovirus humangamma8 | human gammaherpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) | KSHV |
| Transmission Mode | Subfamily | Key Glycoproteins | Host Receptors | Intervention Strategies (Include Under Development) | Intervention Targets | Refs. |
|---|---|---|---|---|---|---|
| Tunneling Nanotubes (TNTs) | Alphaherpesvirinae (e.g., HSV-1) | gB, gD, gE, US3 kinase | Actin cytoskeleton (Arp2/3 complex), E-cadherin/β-catenin | CK666 inhibitors, PHB1 inhibitors (RocA), Shingrix® vaccine (GlaxoSmithKline, Brentford, UK) | PAK1/2, PHB1, gE, gE/gI | [3,4,5,6,7,8,9,10,11,12,13] |
| Gammaherpesvirinae (e.g., EBV) | BMRF2, BDLF2, gp48/ORF58 | Not explicitly mentioned | EBV mRNA vaccine (WGc-043), liposomal nanovaccinations | gHgL, gB, gp42 | [14,15,16] | |
| Exosomes | Alphaherpesvirinae (e.g., HSV-1) | gB, VP16, ICP5, miRNAs | Rab27a, STING, Sp100A | Shingrix® vaccine (VZV) | gE/gI | [11,12,13,17,18,19,20,21] |
| Betaherpesvirinae (e.g., HCMV) | gB, UL128L, miRNAs, | ESCRT proteins | HCMV mRNA (mRNA-1647) | gB and pentameric subunits (gH/gL/UL128/130/131A) | [22,23] | |
| Gammaherpesvirinae (e.g., EBV) | EBERs, miR-BART3, miR-BHRF1-1 | DCs, IFN | EBV mRNA vaccine (WGc-043), liposomal nanovaccinations | gHgL, gB, gp42 | [16,24,25] | |
| Tight Junctions (TJs) | Alphaherpesvirinae (e.g., HSV-1,VZV) | gE, gI, gM, UL7, UL51, VP22, UL56, US7, ICP0 | TGN, Nectin-1, Nectin-2 | PHB1) inhibitors (RocA), BBI inhibitors, IL-22 therapy, Nelfinavir (NFV), Shingrix® vaccine (VZV) | End1/E6E7, ZO-1, OCLN, TGN, gE/gI | [10,11,12,13,26,27,28,29,30,31,32,33,34,35,36,37,38,39] |
| Gammaherpesvirinae (e.g., EBV, KSHV) | gH/gL | EphA2 receptor | EBV mRNA vaccine (WGc-043), liposomal nanovaccinations | gHgL, gB, gp42 | [16,40] | |
| Syncytium | Alphaherpesvirinae (e.g., HSV-1, VZV) | gB, gD (VZV exclusive), gH/gL, gE, gK, gJ, UL16, UL20, UL24 | PTP1B, HVEM, HSPG | PTP1B inhibitors, PHB1 inhibitors (RocA). mAb: m27f, 4A3, HDIT101, HDIT10. Shingrix® vaccine (VZV) | PTP1B, gD (residues 292–297, 216–220), gB, gE/gI | [10,11,41,42,43,44,45,46,47,48,49,50,51] |
| Betaherpesvirinae (e.g., HCMV) | gH/gL, gB, UL128L | Not explicitly mentioned | HCMV mRNA (mRNA-1647) | gB and pentameric subunits (gH/gL/UL128/130/131A) | [23,52,53] | |
| Gammaherpesvirinae (e.g., EBV, KSHV) | Immediate-early gene products | Raji cells, specific receptors | mAb: 3A3, 3A5. EBV mRNA vaccine (WGc-043), liposomal nanovaccinations | gB, NRP1, gH/gL, gB, gp42 | [16,24,25,54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, L.; Guo, J.; Zhong, Y.; Wei, J.; Wang, Z. Molecular Mechanisms of Cell-to-Cell Transmission in Human Herpesviruses. Viruses 2025, 17, 742. https://doi.org/10.3390/v17060742
Yan L, Guo J, Zhong Y, Wei J, Wang Z. Molecular Mechanisms of Cell-to-Cell Transmission in Human Herpesviruses. Viruses. 2025; 17(6):742. https://doi.org/10.3390/v17060742
Chicago/Turabian StyleYan, Liyuan, Jing Guo, Yinan Zhong, Jiangbo Wei, and Zejun Wang. 2025. "Molecular Mechanisms of Cell-to-Cell Transmission in Human Herpesviruses" Viruses 17, no. 6: 742. https://doi.org/10.3390/v17060742
APA StyleYan, L., Guo, J., Zhong, Y., Wei, J., & Wang, Z. (2025). Molecular Mechanisms of Cell-to-Cell Transmission in Human Herpesviruses. Viruses, 17(6), 742. https://doi.org/10.3390/v17060742