
The COPII Transport Complex Participates in HPV16 Infection


Abstract
1. Introduction
2. Materials and Methods
2.1. Pseudovirus and VLP Production
2.2. Immunofluorescence
2.3. HPV PsV Infection
2.4. Cell Cycle Analysis
2.5. siRNA Transfection
2.6. Western Blot Analysis
2.7. Electron Microscopy
2.8. Crosslinking and Immunoprecipitation
3. Results
3.1. TEM Reveals Extended PsV-Containing Vesicles
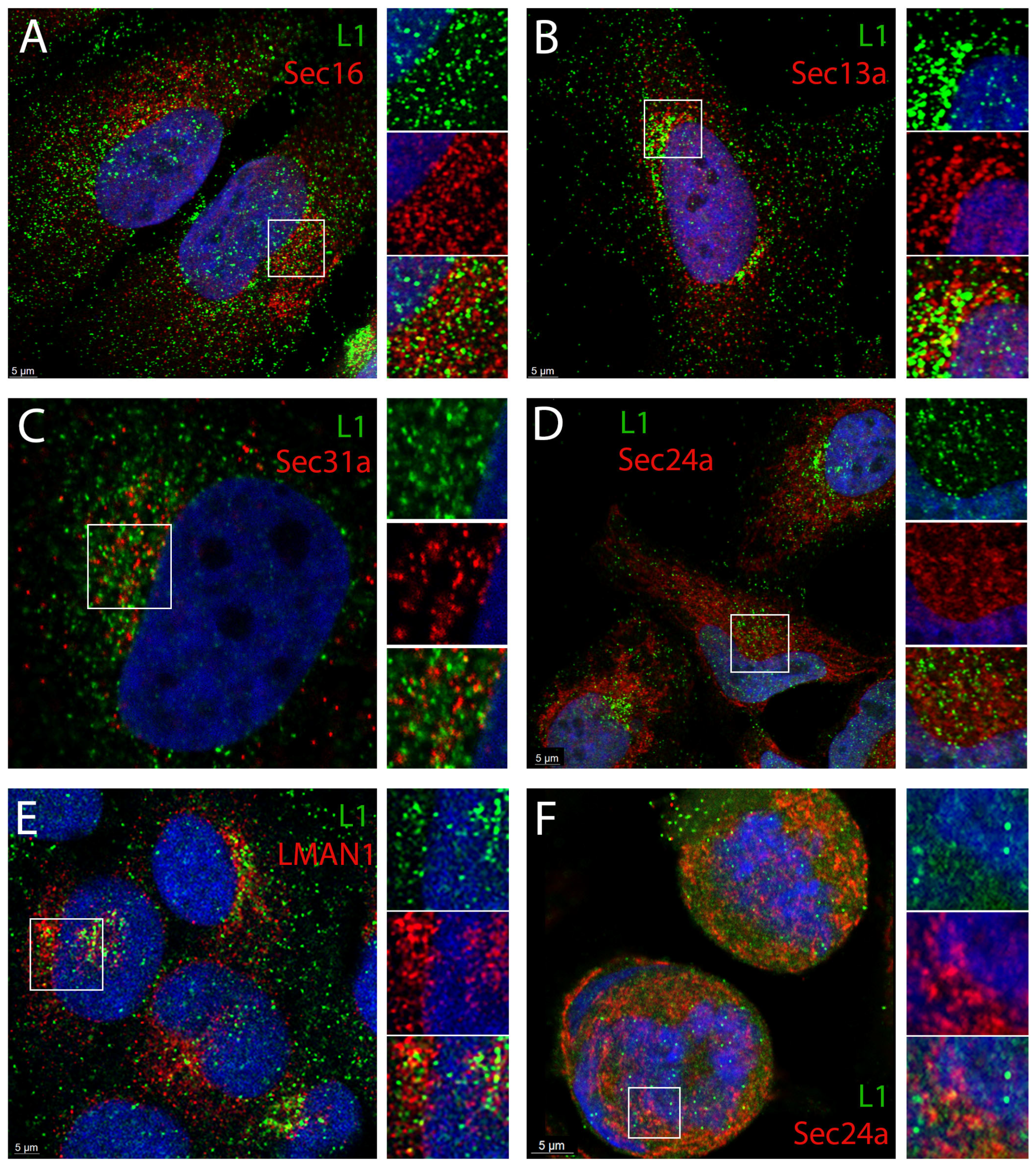
3.2. HPV16 PsV Localize to ERES and COPII Vesicles
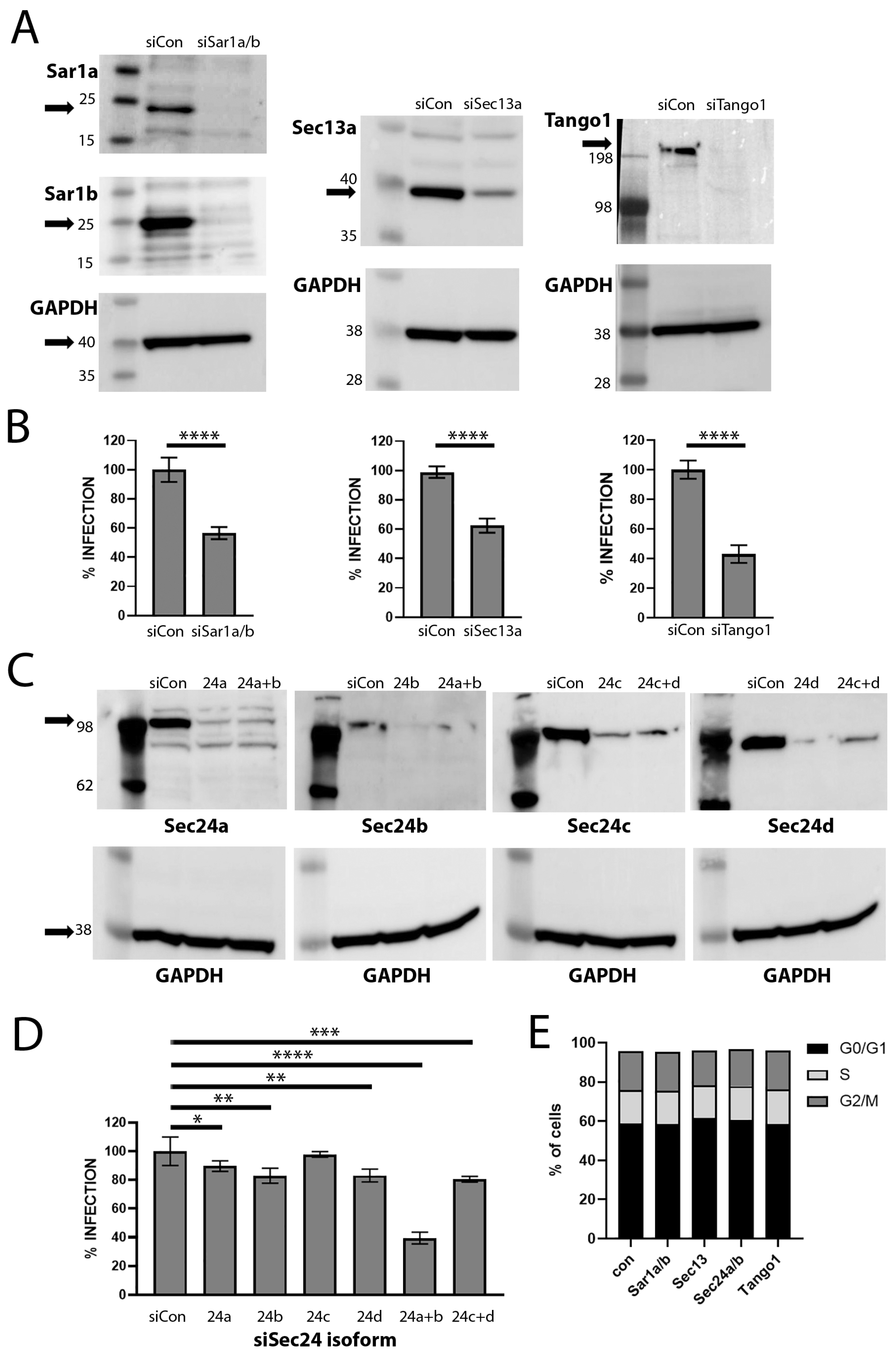
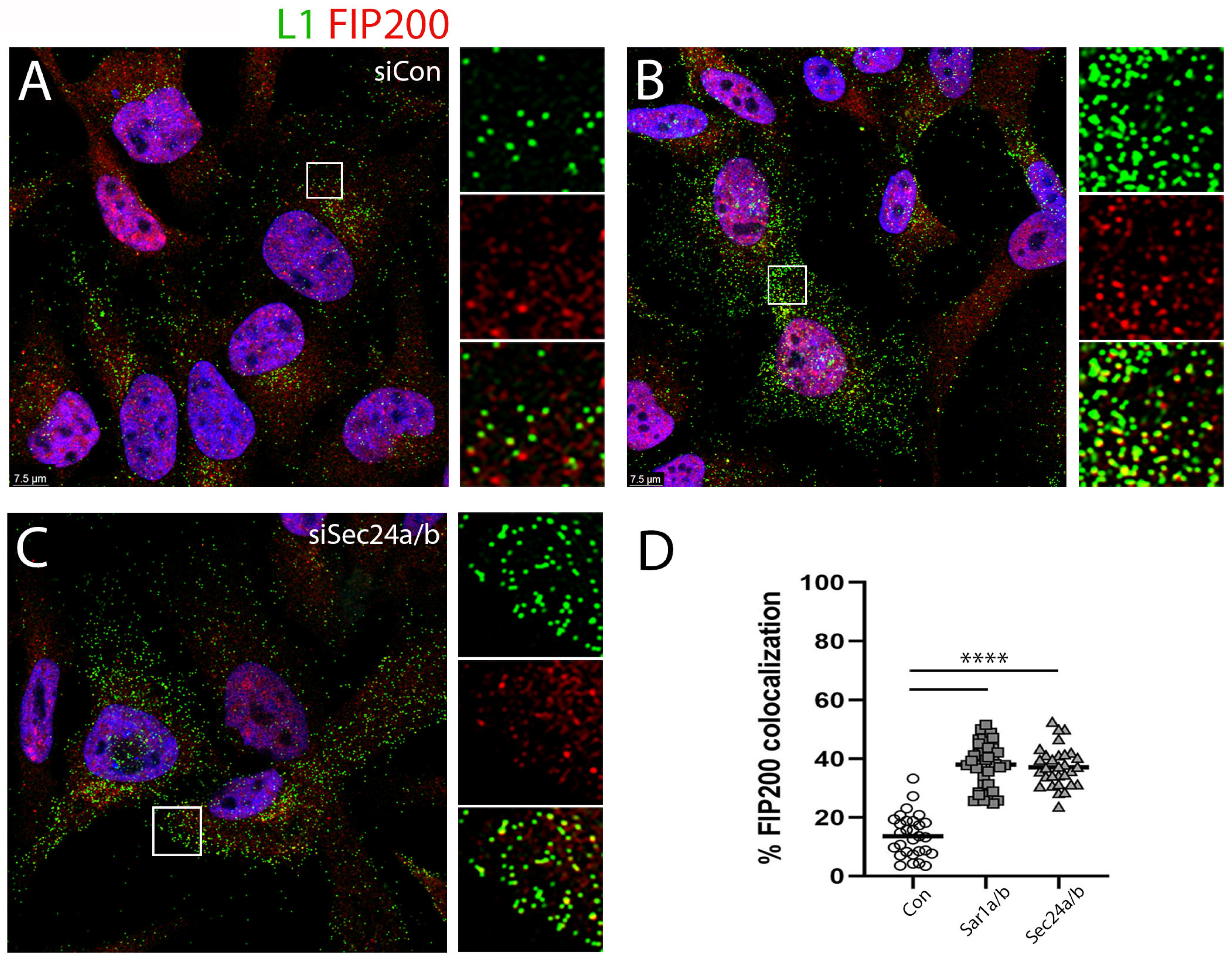
3.3. siRNA Depletion of COPII Transport Mediators Decreases HPV16 PsV Infection
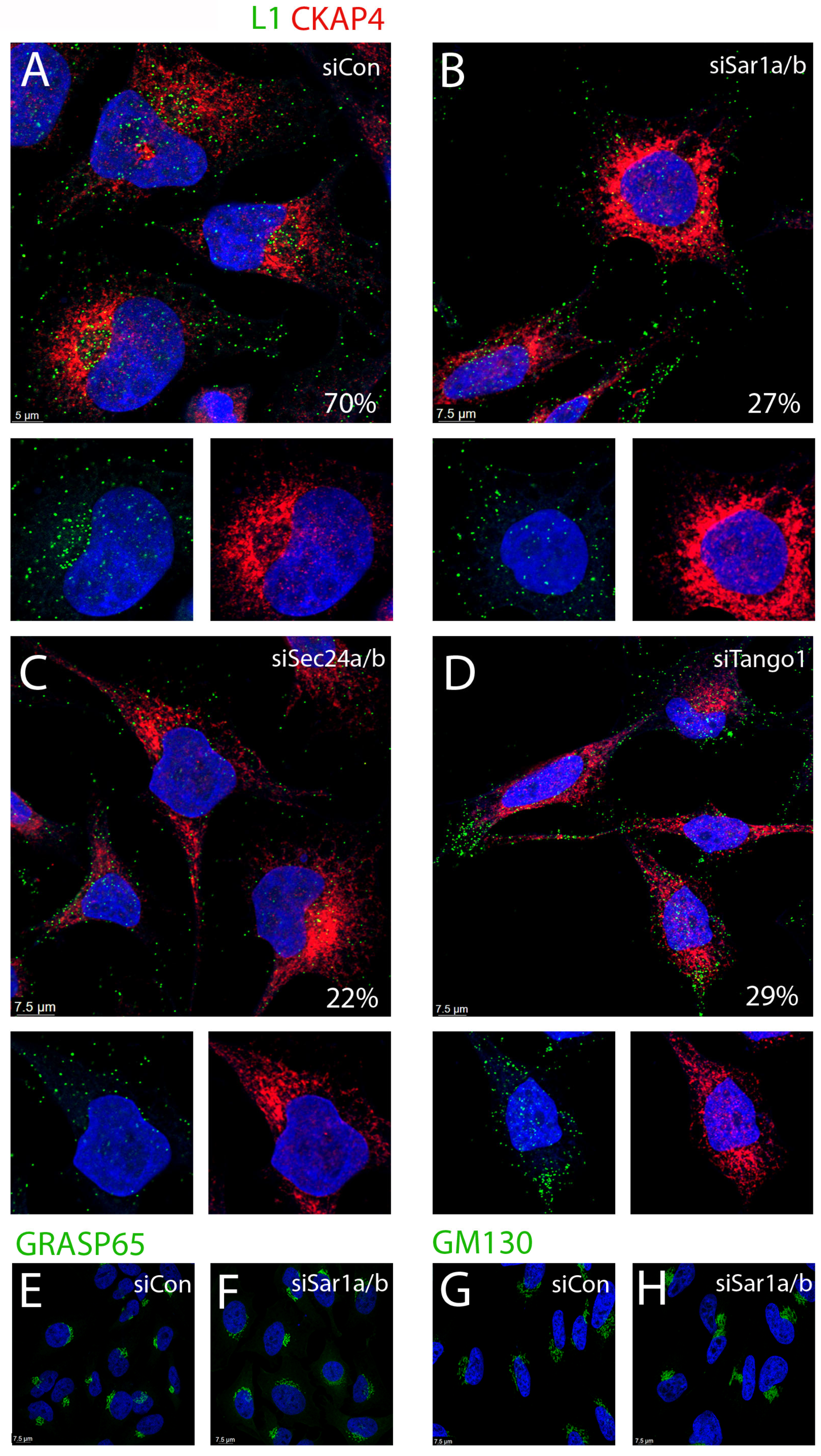
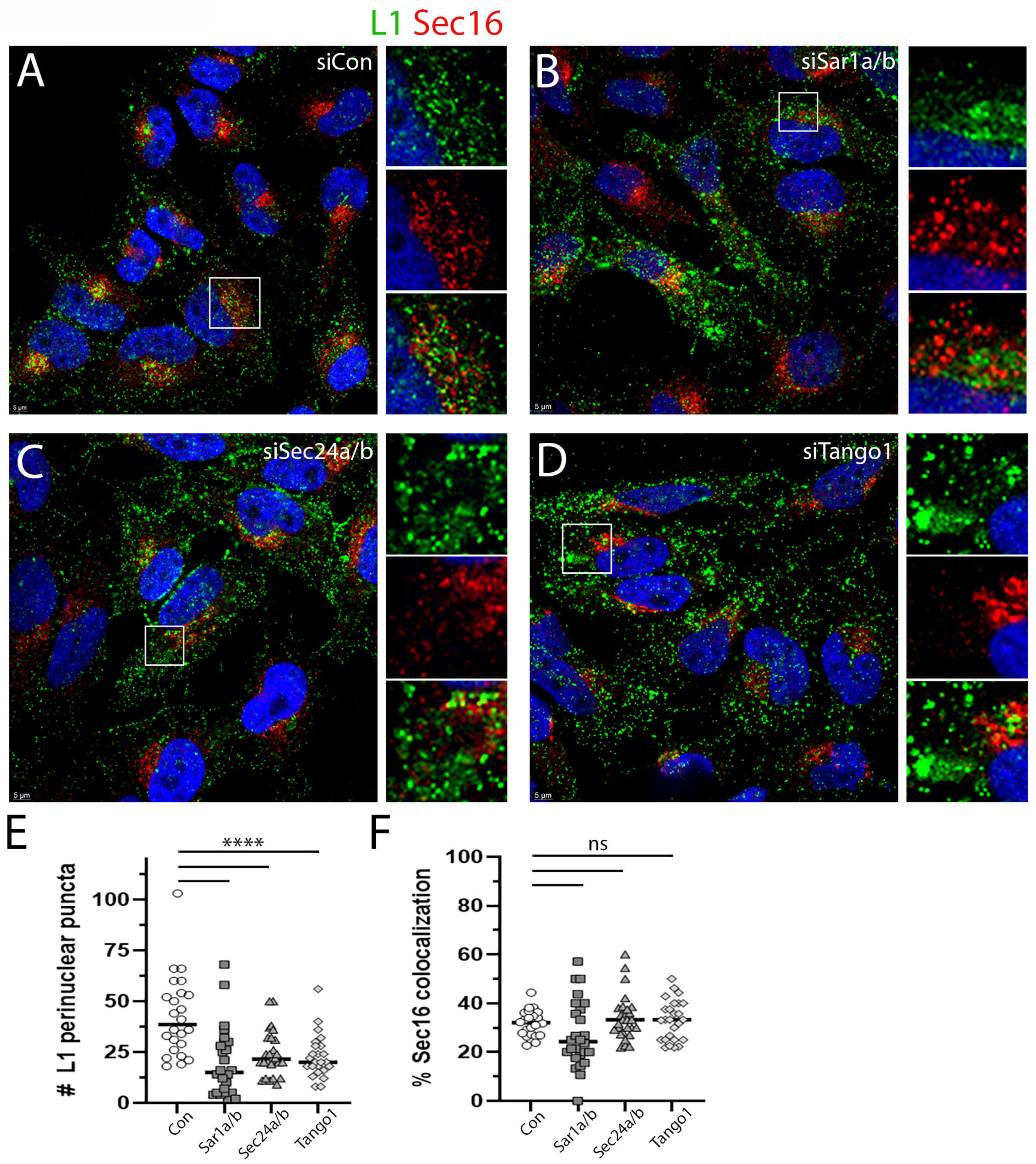
3.4. Depletion of COPII Proteins Affects PsV Trafficking
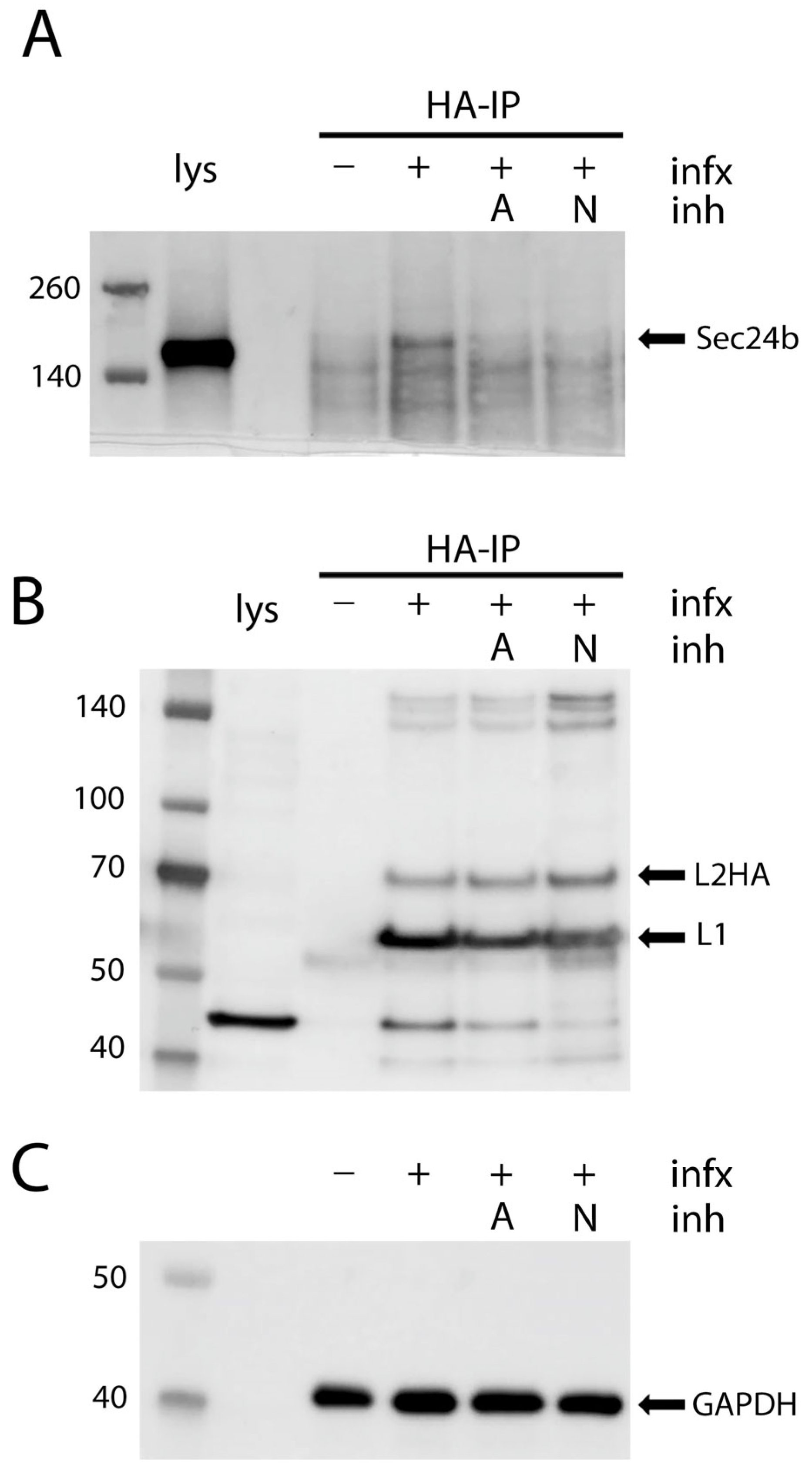
3.5. Co-Immunoprecipitation of COPII Cargo Acceptor with HPV Capsids
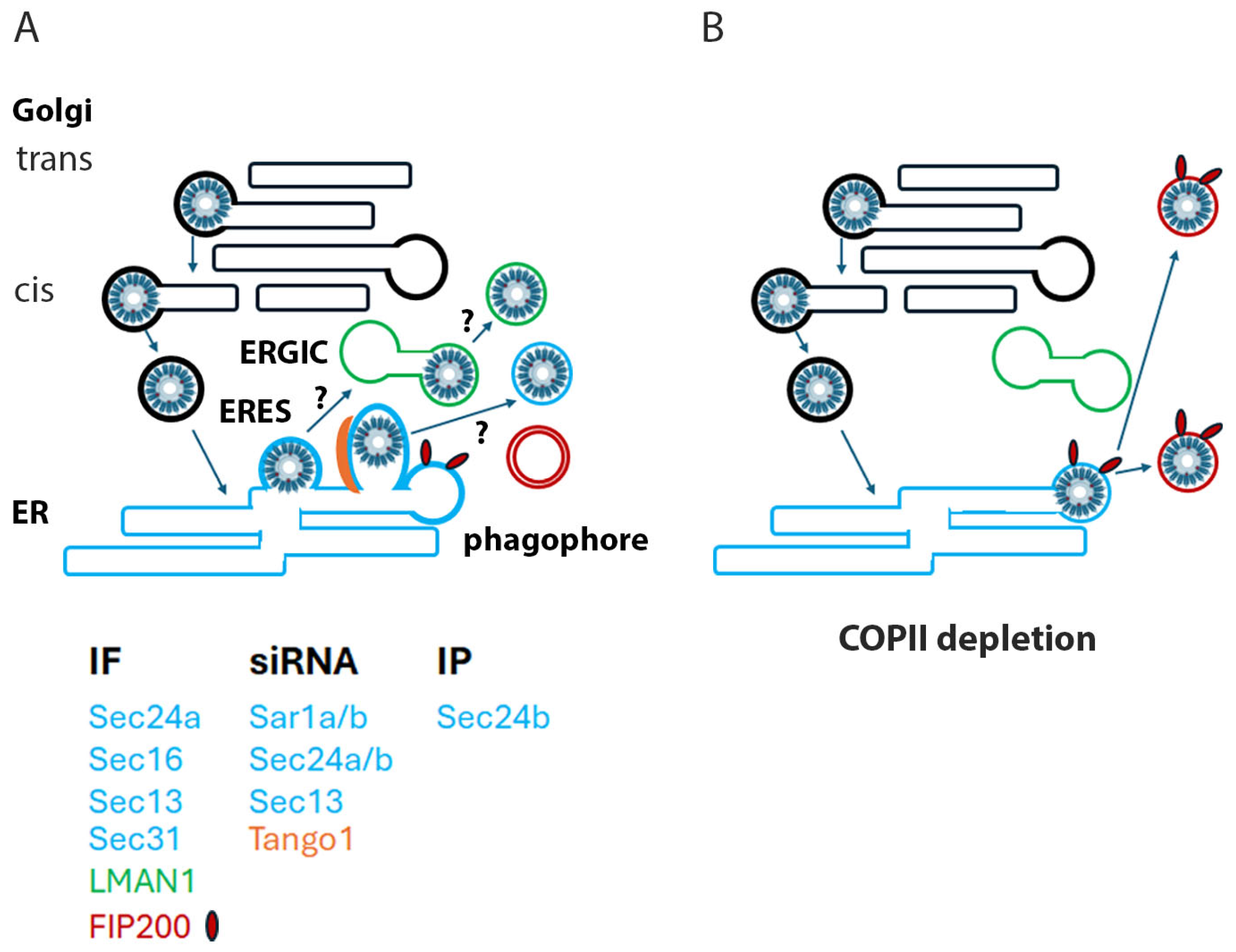
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COPI | coat protein complex I |
| COPII | coat protein complex II |
| ER | Endoplasmic reticulum |
| ERES | ER exit sites |
| ERGIC | ER-Golgi intermediate compartment |
| HPV | Human papillomavirus |
| PsV | Pseudovirus |
| STX18 | Sorting nexin 18 |
| TANGO1 | transport and Golgi organization 1 protein |
| TEM | transmission electron microscopy |
References
- de Martel, C. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Guion, L.; Bienkowska-Haba, M.; DiGiuseppe, S.; Florin, L.; Sapp, M. PML nuclear body-residing proteins sequentially associate with HPV genome after infectious nuclear delivery. PLoS Pathog. 2019, 15, e1007590. [Google Scholar] [CrossRef]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef]
- Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef]
- Inoue, T.; Zhang, P.; Zhang, W.; Goodner-Bingham, K.; Dupzyk, A.; DiMaio, D.; Tsai, B. gamma-Secretase promotes membrane insertion of the human papillomavirus L2 capsid protein during virus infection. J. Cell Biol. 2018, 217, 3545–3559. [Google Scholar] [CrossRef]
- Bossis, I.; Roden, R.B.; Gambhira, R.; Yang, R.; Tagaya, M.; Howley, P.M.; Meneses, P.I. Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J. Virol. 2005, 79, 6723–6731. [Google Scholar] [CrossRef]
- Bergant Marusic, M.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef]
- Broniarczyk, J.; Bergant, M.; Gozdzicka-Jozefiak, A.; Banks, L. Human papillomavirus infection requires the TSG101 component of the ESCRT machinery. Virology 2014, 460–461, 83–90. [Google Scholar] [CrossRef]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Harwood, M.C.; Woo, T.T.; Takeo, Y.; DiMaio, D.; Tsai, B. HPV is a cargo for the COPI sorting complex during virus entry. Sci. Adv. 2023, 9, eadc9830. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M.J. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.H.; Seemann, J. The mitotic spindle mediates inheritance of the Golgi ribbon structure. J. Cell Biol. 2009, 184, 391–397. [Google Scholar] [CrossRef]
- Laniosz, V.; Dabydeen, S.A.; Havens, M.A.; Meneses, P.I. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. J. Virol. 2009, 83, 8221–8232. [Google Scholar] [CrossRef]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio 2014, 5, e01777-14. [Google Scholar] [CrossRef]
- Morante, A.V.; Baboolal, D.D.; Simon, X.; Pan, E.C.; Meneses, P.I. Human Papillomavirus Minor Capsid Protein L2 Mediates Intracellular Trafficking into and Passage beyond the Endoplasmic Reticulum. Microbiol. Spectr. 2022, 10, e0150522. [Google Scholar] [CrossRef] [PubMed]
- Laniosz, V.; Nguyen, K.C.; Meneses, P.I. Bovine papillomavirus type 1 infection is mediated by SNARE syntaxin 18. J. Virol. 2007, 81, 7435–7448. [Google Scholar] [CrossRef]
- Day, P.M.; Weisberg, A.S.; Thompson, C.D.; Hughes, M.M.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Human Papillomavirus 16 Capsids Mediate Nuclear Entry during Infection. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Dodonova, S.O.; Diestelkoetter-Bachert, P.; von Appen, A.; Hagen, W.J.; Beck, R.; Beck, M.; Wieland, F.; Briggs, J.A. VESICULAR TRANSPORT. A structure of the COPI coat and the role of coat proteins in membrane vesicle assembly. Science 2015, 349, 195–198. [Google Scholar] [CrossRef]
- Misteli, T.; Warren, G. COP-coated vesicles are involved in the mitotic fragmentation of Golgi stacks in a cell-free system. J. Cell Biol. 1994, 125, 269–282. [Google Scholar] [CrossRef]
- Sesso, A.; Azimovas, S.R.; Ferreira, M.A. Freeze-fracture and thin section study of the rough ER-Golgi interface in the pancreatic acinar cell. Resemblance between the intramembranal architecture of the outermost Golgi cisterna and the post-rough ER vesicular and tubular elements. Biol. Cell 1994, 81, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Bannykh, S.I.; Rowe, T.; Balch, W.E. The organization of endoplasmic reticulum export complexes. J. Cell Biol. 1996, 135, 19–35. [Google Scholar] [CrossRef]
- Zeuschner, D.; Geerts, W.J.; van Donselaar, E.; Humbel, B.M.; Slot, J.W.; Koster, A.J.; Klumperman, J. Immuno-electron tomography of ER exit sites reveals the existence of free COPII-coated transport carriers. Nat. Cell Biol. 2006, 8, 377–383. [Google Scholar] [CrossRef]
- Nagashima, K.; Zheng, J.; Parmiter, D.; Patri, A.K. Biological tissue and cell culture specimen preparation for TEM nanoparticle characterization. Methods Mol. Biol. 2011, 697, 83–91. [Google Scholar] [CrossRef]
- Santos, A.J.; Raote, I.; Scarpa, M.; Brouwers, N.; Malhotra, V. TANGO1 recruits ERGIC membranes to the endoplasmic reticulum for procollagen export. Elife 2015, 4, e10982. [Google Scholar] [CrossRef]
- Aridor, M.; Bannykh, S.I.; Rowe, T.; Balch, W.E. Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J. Biol. Chem. 1999, 274, 4389–4399. [Google Scholar] [CrossRef] [PubMed]
- Wada, I.; Rindress, D.; Cameron, P.H.; Ou, W.J.; Doherty, J.J., 2nd; Louvard, D.; Bell, A.W.; Dignard, D.; Thomas, D.Y.; Bergeron, J.J. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J. Biol. Chem. 1991, 266, 19599–19610. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Shemesh, T.; Prinz, W.A.; Palazzo, A.F.; Kozlov, M.M.; Rapoport, T.A. Mechanisms determining the morphology of the peripheral ER. Cell 2010, 143, 774–788. [Google Scholar] [CrossRef]
- Paskevicius, T.; Farraj, R.A.; Michalak, M.; Agellon, L.B. Calnexin, More Than Just a Molecular Chaperone. Cells 2023, 12, 403. [Google Scholar] [CrossRef]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Townley, A.K.; Koka, P.; Palmer, K.J.; Stephens, D.J. Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic 2006, 7, 1678–1687. [Google Scholar] [CrossRef]
- Hughes, H.; Budnik, A.; Schmidt, K.; Palmer, K.J.; Mantell, J.; Noakes, C.; Johnson, A.; Carter, D.A.; Verkade, P.; Watson, P.; et al. Organisation of human ER-exit sites: Requirements for the localisation of Sec16 to transitional ER. J. Cell Sci. 2009, 122 Pt 16, 2924–2934. [Google Scholar] [CrossRef]
- Supek, F.; Madden, D.T.; Hamamoto, S.; Orci, L.; Schekman, R. Sec16p potentiates the action of COPII proteins to bud transport vesicles. J. Cell Biol. 2002, 158, 1029–1038. [Google Scholar] [CrossRef]
- Jensen, D.; Schekman, R. COPII-mediated vesicle formation at a glance. J. Cell Sci. 2011, 124 Pt 1, 1–4. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hauri, H.P. The ER-Golgi intermediate compartment (ERGIC): In search of its identity and function. J. Cell Sci. 2006, 119 Pt 11, 2173–2183. [Google Scholar] [CrossRef]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface . Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef]
- Saraste, J.; Palade, G.E.; Farquhar, M.G. Temperature-sensitive steps in the transport of secretory proteins through the Golgi complex in exocrine pancreatic cells. Proc. Natl. Acad. Sci. USA 1986, 83, 6425–6429. [Google Scholar] [CrossRef] [PubMed]
- Saraste, J.; Palade, G.E.; Farquhar, M.G. Antibodies to rat pancreas Golgi subfractions: Identification of a 58-kD cis-Golgi protein. J. Cell Biol. 1987, 105, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Szul, T.; Sztul, E. COPII and COPI traffic at the ER-Golgi interface. Physiology 2011, 26, 348–364. [Google Scholar] [CrossRef]
- Aridor, M.; Fish, K.N.; Bannykh, S.; Weissman, J.; Roberts, T.H.; Lippincott-Schwartz, J.; Balch, W.E. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol. 2001, 152, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Tang, V.T.; Xiang, J.; Chen, Z.; McCormick, J.; Abbineni, P.S.; Chen, X.W.; Hoenerhoff, M.; Emmer, B.T.; Khoriaty, R.; Lin, J.D.; et al. Functional overlap between the mammalian Sar1a and Sar1b paralogs in vivo. Proc. Natl. Acad. Sci. USA 2024, 121, e2322164121. [Google Scholar] [CrossRef]
- Wendeler, M.W.; Paccaud, J.P.; Hauri, H.P. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 2007, 8, 258–264. [Google Scholar] [CrossRef]
- Mancias, J.D.; Goldberg, J. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 2008, 27, 2918–2928. [Google Scholar] [CrossRef]
- Saito, K.; Chen, M.; Bard, F.; Chen, S.; Zhou, H.; Woodley, D.; Polischuk, R.; Schekman, R.; Malhotra, V. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 2009, 136, 891–902. [Google Scholar] [CrossRef]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Ge, L.; Schekman, R. The ER-Golgi intermediate compartment feeds the phagophore membrane. Autophagy 2014, 10, 170–172. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, M.; Kenny, S.J.; Liu, D.; Maeda, M.; Saito, K.; Mathur, A.; Xu, K.; Schekman, R. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017, 18, 1586–1603. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tamura, N.; Kono, N.; Shimanaka, Y.; Arai, H.; Yamamoto, H.; Mizushima, N. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017, 36, 1719–1735. [Google Scholar] [CrossRef]
- Dabydeen, S.A.; Meneses, P.I. The role of NH4Cl and cysteine proteases in Human Papillomavirus type 16 infection. Virol. J. 2009, 6, 109. [Google Scholar] [CrossRef]
- Bisnett, B.J.; Condon, B.M.; Lamb, C.H.; Georgiou, G.R.; Boyce, M. Export Control: Post-transcriptional Regulation of the COPII Trafficking Pathway. Front. Cell Dev. Biol. 2020, 8, 618652. [Google Scholar] [CrossRef]
- Hammond, A.T.; Glick, B.S. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol. Biol. Cell 2000, 11, 3013–3030. [Google Scholar] [CrossRef]
- Bevis, B.J.; Hammond, A.T.; Reinke, C.A.; Glick, B.S. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat. Cell Biol. 2002, 4, 750–756. [Google Scholar] [CrossRef]
- Connerly, P.L.; Esaki, M.; Montegna, E.A.; Strongin, D.E.; Levi, S.; Soderholm, J.; Glick, B.S. Sec16 is a determinant of transitional ER organization. Curr. Biol. 2005, 15, 1439–1447. [Google Scholar] [CrossRef]
- Shindiapina, P.; Barlowe, C. Requirements for transitional endoplasmic reticulum site structure and function in Saccharomyces cerevisiae. Mol. Biol. Cell 2010, 21, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Aridor, M.; Bannykh, S.I.; Rowe, T.; Balch, W.E. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 1995, 131, 875–893. [Google Scholar] [CrossRef]
- Iinuma, T.; Aoki, T.; Arasaki, K.; Hirose, H.; Yamamoto, A.; Samata, R.; Hauri, H.P.; Arimitsu, N.; Tagaya, M.; Tani, K. Role of syntaxin 18 in the organization of endoplasmic reticulum subdomains. J. Cell Sci. 2009, 122 Pt 10, 1680–1690. [Google Scholar] [CrossRef]
- Bard, F.; Casano, L.; Mallabiabarrena, A.; Wallace, E.; Saito, K.; Kitayama, H.; Guizzunti, G.; Hu, Y.; Wendler, F.; Dasgupta, R.; et al. Functional genomics reveals genes involved in protein secretion and Golgi organization. Nature 2006, 439, 604–607. [Google Scholar] [CrossRef]
- Raote, I.; Ortega-Bellido, M.; Santos, A.J.; Foresti, O.; Zhang, C.; Garcia-Parajo, M.F.; Campelo, F.; Malhotra, V. TANGO1 builds a machine for collagen export by recruiting and spatially organizing COPII, tethers and membranes. Elife 2018, 7, e32723. [Google Scholar] [CrossRef]
- Hirata, Y.; Matsui, Y.; Wada, I.; Hosokawa, N. Endoplasmic reticulum-to-Golgi trafficking of procollagen III via conventional vesicular and tubular carriers. Mol. Biol. Cell 2022, 33, ar21. [Google Scholar] [CrossRef]
- Maiers, J.L.; Kostallari, E.; Mushref, M.; deAssuncao, T.M.; Li, H.; Jalan-Sakrikar, N.; Huebert, R.C.; Cao, S.; Malhi, H.; Shah, V.H. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology 2017, 65, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Hirata, Y.; Wada, I.; Hosokawa, N. Visualization of Procollagen IV Reveals ER-to-Golgi Transport by ERGIC-independent Carriers. Cell Struct. Funct. 2020, 45, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Erlmann, P.; Villeneuve, J.; Santos, A.J.; Martinez-Alonso, E.; Martinez-Menarguez, J.A.; Malhotra, V. SLY1 and Syntaxin 18 specify a distinct pathway for procollagen VII export from the endoplasmic reticulum. Elife 2014, 3, e02784. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, P.; Crite, M.; DiMaio, D. Papillomaviruses Go Retro. Pathogens 2020, 9, 267. [Google Scholar] [CrossRef]
- Shomron, O.; Nevo-Yassaf, I.; Aviad, T.; Yaffe, Y.; Zahavi, E.E.; Dukhovny, A.; Perlson, E.; Brodsky, I.; Yeheskel, A.; Pasmanik-Chor, M.; et al. COPII collar defines the boundary between ER and ER exit site and does not coat cargo containers. J. Cell Biol. 2021, 220, e201907224. [Google Scholar] [CrossRef] [PubMed]
- Cutrona, M.B.; Beznoussenko, G.V.; Fusella, A.; Martella, O.; Moral, P.; Mironov, A.A. Silencing of mammalian Sar1 isoforms reveals COPII-independent protein sorting and transport. Traffic 2013, 14, 691–708. [Google Scholar] [CrossRef]
- Liu, J.; Prunuske, A.J.; Fager, A.M.; Ullman, K.S. The COPI complex functions in nuclear envelope breakdown and is recruited by the nucleoporin Nup153. Dev. Cell 2003, 5, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Zaal, K.J.; Smith, C.L.; Polishchuk, R.S.; Altan, N.; Cole, N.B.; Ellenberg, J.; Hirschberg, K.; Presley, J.F.; Roberts, T.H.; Siggia, E.; et al. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell 1999, 99, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.M.; Cicchini, L.; Pyeon, D. Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology 2013, 437, 12–19. [Google Scholar] [CrossRef]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef]
- Otomo, T.; Chowdhury, S.; Lander, G.C. The rod-shaped ATG2A-WIPI4 complex tethers membranes in vitro. Contact 2018, 1, 2515256418819936. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sar1a | #1 GAACAGAUGCAAUCAGUGA, #2 CCAGUAUAUUGACUGAUGU (Sigma) |
| Sar1b | #1 GCAUAACUUGAAUUCAAUA, #2 CUACCUUCCUGCUAUCAAU (Sigma) |
| Sec13 | #1 CAUGUGAGCUGGUCCAUCA, #2 GGUCGUGUGUUCAUUUGGA, #3 CCAUCUCCCUGCUGACUUA, #4 GUAAUUAACACUGUGGAUA (Dharmacon) |
| Sec24a | #1 GGACGUACAUCAAUCCUUU, #2 CCAAGAAGGUAUUACAUCA, #3 GUGGUUACCUCCAGUACAA, #4 CAGUAGUUACGACGAGAUU (Dharmacon) |
| Sec24b | #1 CUUCAGAGACCUAACGCAA, #2 CCAGAUUCAUUUCGGUGUA, #3 CUUCAUUGAUCAACGUAGA, #4 GCUAUAGAGUAAACGAUGU (Dharmacon) |
| Sec24c | #1 GCACAGAGAUCCCGGUACA, #2 UGGCUGAUCUAUAUCGAAA, #3 CCUUUCAGGUGGAGAACGA, #4 CCUGGAUCAUACCGGCAAA (Dharmacon) |
| Sec24d | #1 GGUAAAUCACGGCGAGAGU, #2 GAUCUCAACUGAUGAACGA, #3 UUGAAGGUCAUCCGGGAAA, #4 CGUUAGAUGUCAAGAGUAC (Dharmacon) |
| TANGO1 | GAUAAGGUCUUCCGUGCUU (Sigma) |
| Universal negative control | predesigned (Sigma #SIC001) |
| Target (Name) | Usage (Dilution) | Source (Catalog #) |
|---|---|---|
| CKAP4 | IF (1/800) | Proteintech (16686-1-AP) |
| GAPDH | WB (1/1000) | Novus (NBP2-27103) |
| HA Tag | IP (4 μg/sample) | Epicypher (13-2010) |
| HA Tag | WB (1/100) | Santa Cruz Biotechnology (F-7) |
| HPV16 L1 (H16.7E) | IF (1/300) | Neil Christensen |
| HPV16 L1 (Camvir-1) | WB (1/10,000) | Abcam (ab69) |
| FIP200 | IF (1/400) | Abcam (ab227726) |
| GM130 | IF (1/200) | BD Biosciences (610822) |
| GRASP65 | IF (1/500) | Invitrogen (PA3-910) |
| LMAN1 | IF (1/200) | Novus (NBP3-04910) |
| Sar1A | WB 1/1000 | Novus (NBP2-20261) |
| Sar1B | WB 1/1000 | Novus (NBP1-32725) |
| Sec13 | WB (1/1000) | Invitrogen (PA5-21339) |
| Sec16A | IF (1/300) | Invitrogen (PA5-52182) |
| Sec24A | IF (1/500) | Proteintech (15958-1-AP) |
| WB (1/2000) | ||
| Sec24B | WB (1/1000) | Cell Signaling Tech (12042) |
| Sec24C | WB (1/1000) | Cell Signaling Tech (14676) |
| Sec24D | WB (1/1000) | Cell Signaling Tech (14687) |
| Sec31A | IF (1/300) | Abcam (ab86600) |
| WB 1/1000 | ||
| TANGO1/MIA3 | WB (1/1000) | Abcam (ab244506) |
| Donkey anti-mouse IgG Alexa Fluor 488 | ||
| IF (1/1000) | Invitrogen (A32766) | |
| Donkey anti-rabbit IgG Alexa Fluor 594 | ||
| IF (1/1000) | Invitrogen (A21207) | |
| Goat anti-mouse IgG HRP | ||
| WB (1/5000) | Invitrogen (A16072) | |
| Goat anti-rabbit IgG HRP | ||
| WB (1/5000) | Invitrogen (A16110) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, P.M.; Thompson, C.D.; Weisberg, A.S.; Schiller, J.T. The COPII Transport Complex Participates in HPV16 Infection. Viruses 2025, 17, 616. https://doi.org/10.3390/v17050616
Day PM, Thompson CD, Weisberg AS, Schiller JT. The COPII Transport Complex Participates in HPV16 Infection. Viruses. 2025; 17(5):616. https://doi.org/10.3390/v17050616
Chicago/Turabian StyleDay, Patricia M., Cynthia D. Thompson, Andrea S. Weisberg, and John T. Schiller. 2025. "The COPII Transport Complex Participates in HPV16 Infection" Viruses 17, no. 5: 616. https://doi.org/10.3390/v17050616
APA StyleDay, P. M., Thompson, C. D., Weisberg, A. S., & Schiller, J. T. (2025). The COPII Transport Complex Participates in HPV16 Infection. Viruses, 17(5), 616. https://doi.org/10.3390/v17050616