
Is the vIL-10 Protein from Cytomegalovirus Associated with the Potential Development of Acute Lymphoblastic Leukemia?
 ,
,  , , ,
, , ,  , , ,
, , , 
Abstract
1. Introduction
2. hCMV and Leukemogenesis
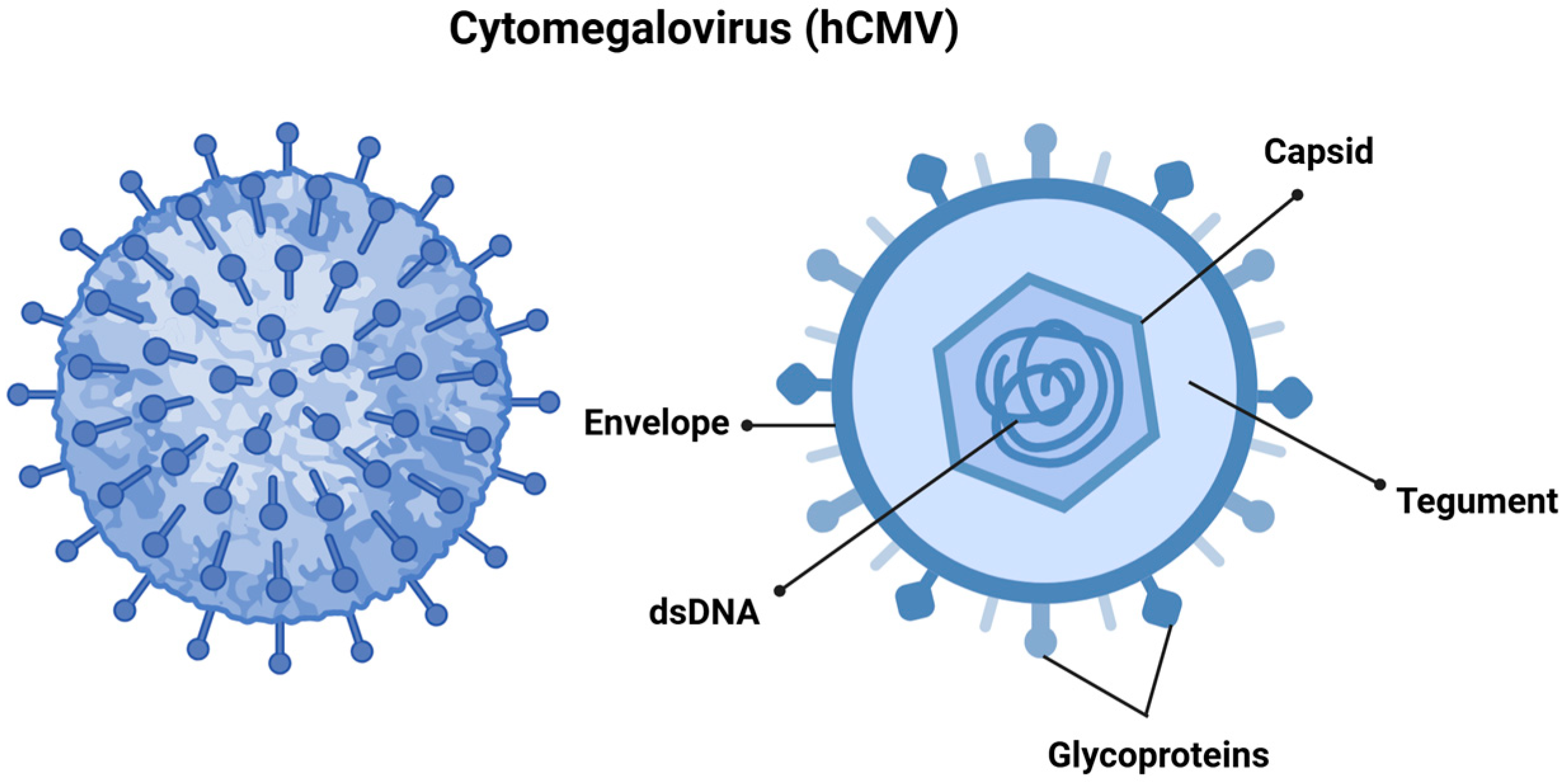
2.1. Human Cytomegalovirus
2.2. Epidemiology
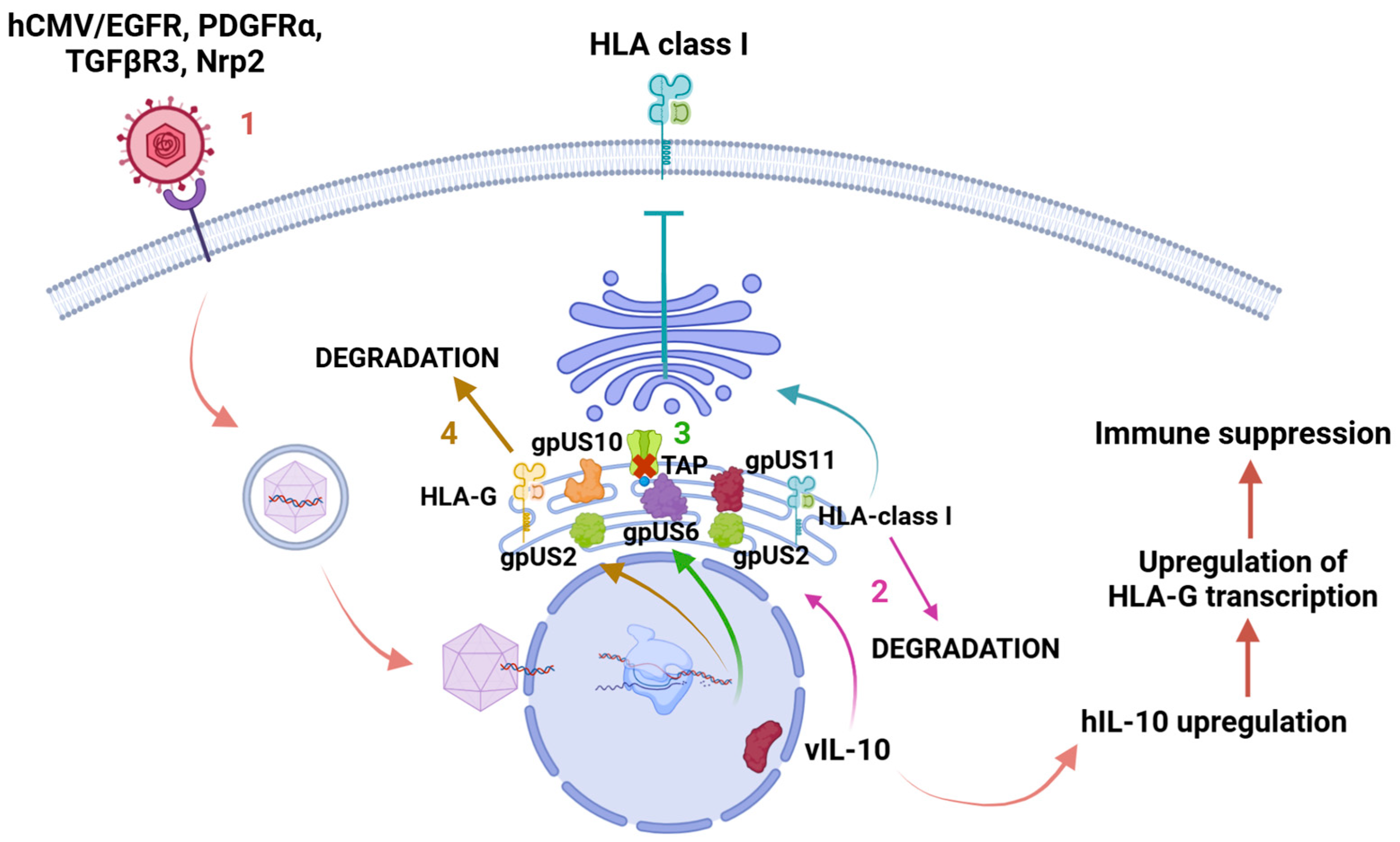
2.3. Role of Immune Evasion of hCMV in Tumorigenesis
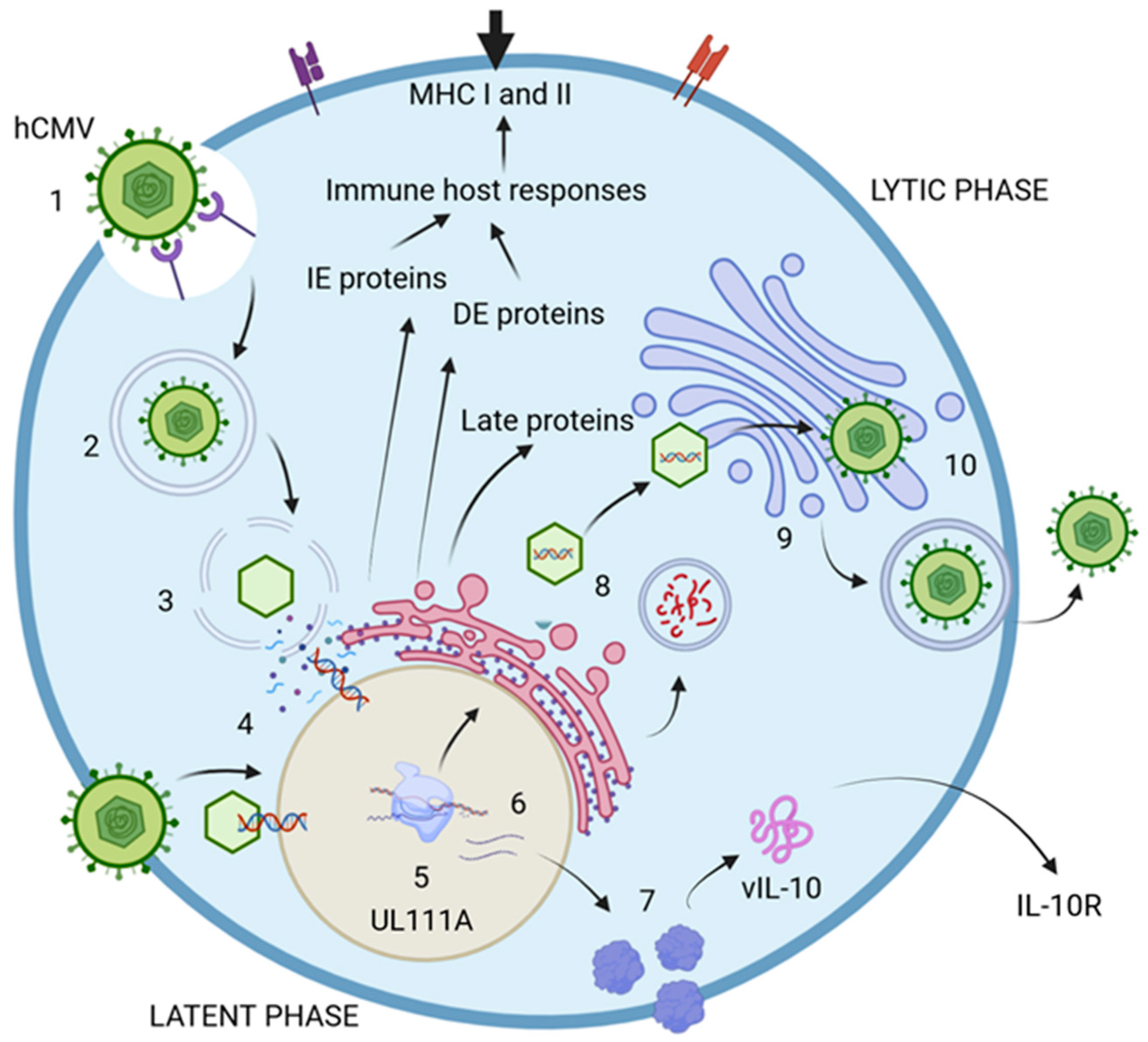
2.4. hCMV Viral Life Cycle and the Latency Proteins
2.5. Viral Infection and Malignance
2.5.1. Oncogenic Viruses
2.5.2. A Comparison Between hCMV and Other Oncogenic Viruses
2.5.3. hCMV VIL10 and Malignance
2.6. IL-10 and vIL10 in hCMV Infection
3. The Role of the Immune System in Tumorigenesis
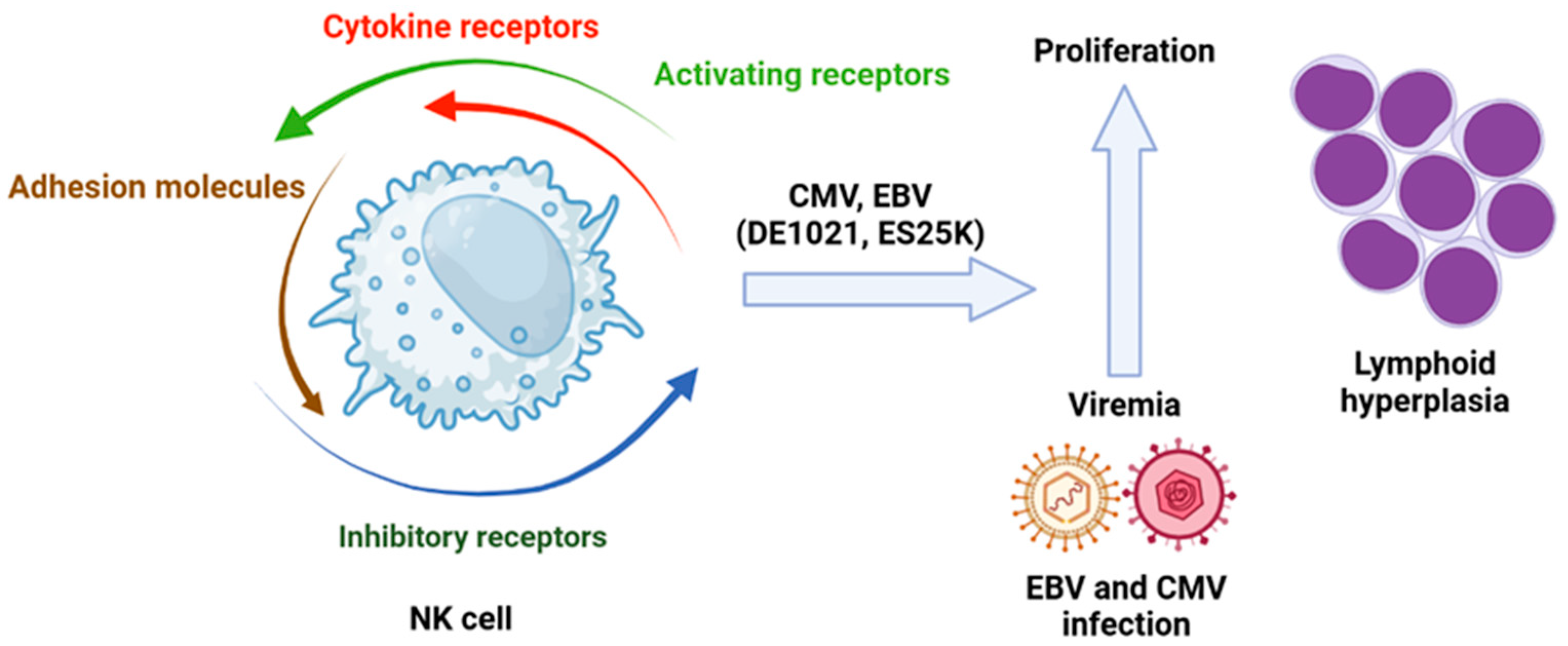
4. The Failure of NK Cells and Tumorigenesis
5. The Influence of Altered Macrophages in the Leukemic Microenviroment
6. Influence of the Altered Acquired Immune System in the Leukemic Microenvironment
7. The Antileukemic Potential of hCMV
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, J.; Chan, S.C.; Ngai, C.H.; Lok, V.; Zhang, L.; Lucero-Prisno, D.E.; Xu, W.; Zheng, Z.-J.; Elcarte, E.; Withers, M.; et al. Disease Burden, Risk Factors, and Trends of Leukaemia: A Global Analysis. Front. Oncol. 2022, 12, 904292. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.; Abdul-Hay, M. Acute Lymphoblastic Leukemia: A Comprehensive Review and 2017 Update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Puckett, Y.; Chan, O. Acute Lymphocytic Leukemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Hamilton, S.T.; Scott, G.; Naing, Z.; Iwasenko, J.; Hall, B.; Graf, N.; Arbuckle, S.; Craig, M.E.; Rawlinson, W.D. Human Cytomegalovirus-Induces Cytokine Changes in the Placenta with Implications for Adverse Pregnancy Outcomes. PLoS ONE 2012, 7, e52899. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.W.; Baumgarth, N.; Yu, D.; Barry, P.A. Human Cytomegalovirus-Encoded Interleukin-10 Homolog Inhibits Maturation of Dendritic Cells and Alters Their Functionality. J. Virol. 2004, 78, 8720–8731. [Google Scholar] [CrossRef]
- Christiaansen, A.; Varga, S.M.; Spencer, J.V. Viral Manipulation of the Host Immune Response. Curr. Opin. Immunol. 2015, 36, 54–60. [Google Scholar] [CrossRef]
- Greaves, M. The ‘Delayed Infection’ (Aka ‘Hygiene’) Hypothesis for Childhood Leukaemia. In The Hygiene Hypothesis and Darwinian Medicine; Rook, G.A.W., Ed.; Birkhäuser Basel: Basel, Switzerland, 2009; pp. 239–255. ISBN 978-3-7643-8902-4. [Google Scholar]
- Gupta, M.; Shorman, M. Cytomegalovirus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Nelson, C.T.; Demmler, G.J. Cytomegalovirus Infection in the Pregnant Mother, Fetus, and Newborn Infant. Clin. Perinatol. 1997, 24, 151–160. [Google Scholar] [CrossRef]
- Pass, R.F. Cytomegalovirus Infection. Pediatr. Rev. 2002, 23, 163–170. [Google Scholar] [CrossRef]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the Worldwide Seroprevalence of Cytomegalovirus: A Systematic Review and Meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Kenneson, A.; Cannon, M.J. Review and Meta-analysis of the Epidemiology of Congenital Cytomegalovirus (CMV) Infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Lagasse, N.; Dhooge, I.; Govaert, P. Congenital CMV-Infection and Hearing Loss. Acta Otorhinolaryngol. Belg. 2000, 54, 431–436. [Google Scholar]
- Leung, A.K.C.; Sauve, R.S.; Davies, H.D. Congenital Cytomegalovirus Infection. J. Natl. Med. Assoc. 2003, 95, 213–218. [Google Scholar] [PubMed]
- Malm, G.; Engman, M.-L. Congenital Cytomegalovirus Infections. Semin. Fetal Neonatal Med. 2007, 12, 154–159. [Google Scholar] [CrossRef]
- Stagno, S.; Pass, R.F.; Dworsky, M.E.; Henderson, R.E.; Moore, E.G.; Walton, P.D.; Alford, C.A. Congenital Cytomegalovirus Infection: The Relative Importance of Primary and Recurrent Maternal Infection. N. Engl. J. Med. 1982, 306, 945–949. [Google Scholar] [CrossRef]
- Söderberg-Nauclér, C. Does Cytomegalovirus Play a Causative Role in the Development of Various Inflammatory Diseases and Cancer? J. Intern. Med. 2006, 259, 219–246. [Google Scholar] [CrossRef]
- Wiemels, J.L.; Wang, R.; Zhou, M.; Hansen, H.; Gallant, R.; Jung, J.; Mancuso, N.; De Smith, A.J.; Metayer, C.; Kogan, S.C.; et al. Cytomegalovirus Proteins, Maternal Pregnancy Cytokines, and Their Impact on Neonatal Immune Cytokine Profiles and Acute Lymphoblastic Leukemogenesis in Children. Haematologica 2022, 107, 2266–2270. [Google Scholar] [CrossRef]
- Gardner, T.J.; Tortorella, D. Virion Glycoprotein-Mediated Immune Evasion by Human Cytomegalovirus: A Sticky Virus Makes a Slick Getaway. Microbiol. Mol. Biol. Rev. 2016, 80, 663–677. [Google Scholar] [CrossRef]
- Ahn, K.; Angulo, A.; Ghazal, P.; Peterson, P.A.; Yang, Y.; Früh, K. Human Cytomegalovirus Inhibits Antigen Presentation by a Sequential Multistep Process. Proc. Natl. Acad. Sci. USA 1996, 93, 10990–10995. [Google Scholar] [CrossRef]
- Jones, T.R.; Wiertz, E.J.; Sun, L.; Fish, K.N.; Nelson, J.A.; Ploegh, H.L. Human Cytomegalovirus US3 Impairs Transport and Maturation of Major Histocompatibility Complex Class I Heavy Chains. Proc. Natl. Acad. Sci. USA 1996, 93, 11327–11333. [Google Scholar] [CrossRef]
- Noriega, V.M.; Tortorella, D. Human Cytomegalovirus-Encoded Immune Modulators Partner To Downregulate Major Histocompatibility Complex Class I Molecules. J. Virol. 2009, 83, 1359–1367. [Google Scholar] [CrossRef]
- Hegde, N.R.; Tomazin, R.A.; Wisner, T.W.; Dunn, C.; Boname, J.M.; Lewinsohn, D.M.; Johnson, D.C. Inhibition of HLA-DR Assembly, Transport, and Loading by Human Cytomegalovirus Glycoprotein US3: A Novel Mechanism for Evading Major Histocompatibility Complex Class II Antigen Presentation. J. Virol. 2002, 76, 10929–10941. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.H.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The Human Cytomegalovirus US11 Gene Product Dislocates MHC Class I Heavy Chains from the Endoplasmic Reticulum to the Cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Shamu, C.E.; Flierman, D.; Ploegh, H.L.; Rapoport, T.A.; Chau, V. Polyubiquitination Is Required for US11-Dependent Movement of MHC Class I Heavy Chain from Endoplasmic Reticulum into Cytosol. MBoC 2001, 12, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Tomazin, R.; Boname, J.; Hegde, N.R.; Lewinsohn, D.M.; Altschuler, Y.; Jones, T.R.; Cresswell, P.; Nelson, J.A.; Riddell, S.R.; Johnson, D.C. Cytomegalovirus US2 Destroys Two Components of the MHC Class II Pathway, Preventing Recognition by CD4+ T Cells. Nat. Med. 1999, 5, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Gewurz, B.E.; Wang, E.W.; Tortorella, D.; Schust, D.J.; Ploegh, H.L. Human Cytomegalovirus US2 Endoplasmic Reticulum-Lumenal Domain Dictates Association with Major Histocompatibility Complex Class I in a Locus-Specific Manner. J. Virol. 2001, 75, 5197–5204. [Google Scholar] [CrossRef]
- Furman, M.H.; Dey, N.; Tortorella, D.; Ploegh, H.L. The Human Cytomegalovirus US10 Gene Product Delays Trafficking of Major Histocompatibility Complex Class I Molecules. J. Virol. 2002, 76, 11753–11756. [Google Scholar] [CrossRef]
- Park, B.; Spooner, E.; Houser, B.L.; Strominger, J.L.; Ploegh, H.L. The HCMV Membrane Glycoprotein US10 Selectively Targets HLA-G for Degradation. J. Exp. Med. 2010, 207, 2033–2041. [Google Scholar] [CrossRef]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T.R.; Wiertz, E.J.H.J.; Ploegh, H.L.; Peterson, P.A.; Yang, Y.; Früh, K. The ER-Luminal Domain of the HCMV Glycoprotein US6 Inhibits Peptide Translocation by TAP. Immunity 1997, 6, 613–621. [Google Scholar] [CrossRef]
- Lehner, P.J.; Karttunen, J.T.; Wilkinson, G.W.G.; Cresswell, P. The Human Cytomegalovirus US6 Glycoprotein Inhibits Transporter Associated with Antigen Processing-Dependent Peptide Translocation. Proc. Natl. Acad. Sci. USA 1997, 94, 6904–6909. [Google Scholar] [CrossRef]
- Trgovcich, J.; Cebulla, C.; Zimmerman, P.; Sedmak, D.D. Human Cytomegalovirus Protein Pp71 Disrupts Major Histocompatibility Complex Class I Cell Surface Expression. J. Virol. 2006, 80, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Abate, D.A.; Watanabe, S.; Mocarski, E.S. Major Human Cytomegalovirus Structural Protein Pp65 (ppUL83) Prevents Interferon Response Factor 3 Activation in the Interferon Response. J. Virol. 2004, 78, 10995–11006. [Google Scholar] [CrossRef] [PubMed]
- Kaarbø, M.; Ager-Wick, E.; Osenbroch, P.Ø.; Kilander, A.; Skinnes, R.; Müller, F.; Eide, L. Human Cytomegalovirus Infection Increases Mitochondrial Biogenesis. Mitochondrion 2011, 11, 935–945. [Google Scholar] [CrossRef]
- Longdon, B.; Brockhurst, M.A.; Russell, C.A.; Welch, J.J.; Jiggins, F.M. The Evolution and Genetics of Virus Host Shifts. PLoS Pathog. 2014, 10, e1004395. [Google Scholar] [CrossRef]
- Slobedman, B.; Mocarski, E.S.; Arvin, A.M.; Mellins, E.D.; Abendroth, A. Latent Cytomegalovirus Down-Regulates Major Histocompatibility Complex Class II Expression on Myeloid Progenitors. Blood 2002, 100, 2867–2873. [Google Scholar] [CrossRef]
- Roetman, J.J.; Apostolova, M.K.I.; Philip, M. Viral and Cellular Oncogenes Promote Immune Evasion. Oncogene 2022, 41, 921–929. [Google Scholar] [CrossRef]
- Herbein, G. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses 2018, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- El Baba, R.; Herbein, G. Immune Landscape of CMV Infection in Cancer Patients: From “Canonical” Diseases Toward Virus-Elicited Oncomodulation. Front. Immunol. 2021, 12, 730765. [Google Scholar] [CrossRef]
- Dutko, F.; Oldstone, M. Cytomegalovirus Causes a Latent Infection in Undifferentiated Cells and Is Activated by Induction of Cell Differentiation. J. Exp. Med. 1981, 154, 1636–1651. [Google Scholar] [CrossRef]
- Jean Beltran, P.M.; Cristea, I.M. The Life Cycle and Pathogenesis of Human Cytomegalovirus Infection: Lessons from Proteomics. Expert Rev. Proteom. 2014, 11, 697–711. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Emery, V.C.; Milne, R. Cytomegalovirus. In Clinical Virology; Richman, D.D., Whitley, R.J., Hayden, F.G., Eds.; Wiley: Hoboken, NJ, USA, 2009; p. 475. ISBN 978-1-68367-407-8. [Google Scholar]
- Isomura, H.; Stinski, M.F.; Murata, T.; Yamashita, Y.; Kanda, T.; Toyokuni, S.; Tsurumi, T. The Human Cytomegalovirus Gene Products Essential for Late Viral Gene Expression Assemble into Prereplication Complexes before Viral DNA Replication. J. Virol. 2011, 85, 6629–6644. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, R.M.; Depto, A.S.; Fortney, J.; Nelson, J.A. Regulated Expression of Early and Late RNAs and Proteins from the Human Cytomegalovirus Immediate-Early Gene Region. J. Virol. 1989, 63, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, T.; Fons, M.P.; Boldogh, I.; AbuBakar, S.; Deng, C.Z.; Millinoff, D. Metabolic and Cellular Effects of Human Cytomegalovirus Infection. Transplant. Proc. 1991, 23, 48–54, discussion 54–55. [Google Scholar]
- Cheung, A.K.L.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral Gene Expression during the Establishment of Human Cytomegalovirus Latent Infection in Myeloid Progenitor Cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A Novel Viral Transcript with Homology to Human Interleukin-10 Is Expressed during Latent Human Cytomegalovirus Infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Damian, D. The Role of Viruses in Cellular Transformation and Cancer. Cancer Rep. 2025, 8, e70150. [Google Scholar] [CrossRef]
- Yee, T.M.; Wang, L.W. Metabolic Reprogramming in Epstein–Barr Virus Associated Diseases. J. Med. Virol. 2025, 97, e70197. [Google Scholar] [CrossRef]
- Gómez-Archila, J.; Arellano-Galindo, J.; Palacios-Reyes, C.; Espinosa-García, A.; Alonso-Themann, P.; Xicohtencatl-Cortes, J.; Ochoa, S.; Cruz-Córdova, A.; Palma-Lara, I. Epstein-Barr Virus as a Promoter of Tumorigenesis in the Tumor Microenvironment of Breast Cancer (Review). Int. J. Mol. Med. 2023, 52, 72. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, L.; Zuo, L.; Gao, S.; Jiang, X.; Han, Y.; Lin, J.; Peng, M.; Wu, N.; Tang, Y.; et al. HPV E6/E7: Insights into Their Regulatory Role and Mechanism in Signaling Pathways in HPV-Associated Tumor. Cancer Gene Ther. 2024, 31, 9–17. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Y.; Feng, X.; Wang, X.; Wu, M.; Gong, L.; Shu, B.; Lu, Q.; Dong, J. HBx Acts as an Oncogene and Promotes the Invasion and Metastasis of Hepatocellular Carcinoma Both in Vivo and Vitro. Dig. Liver Dis. 2021, 53, 360–366. [Google Scholar] [CrossRef]
- Deng, L.; Nagano-Fujii, M.; Tanaka, M.; Nomura-Takigawa, Y.; Ikeda, M.; Kato, N.; Sada, K.; Hotta, H. NS3 Protein of Hepatitis C Virus Associates with the Tumour Suppressor P53 and Inhibits Its Function in an NS3 Sequence-Dependent Manner. J. Gen. Virol. 2006, 87, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and Molecular Mechanisms of Renal Fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Li, S.; Xie, Y.; Yu, C.; Zheng, C.; Xu, Z. The Battle between Host Antiviral Innate Immunity and Immune Evasion by Cytomegalovirus. Cell. Mol. Life Sci. 2024, 81, 341. [Google Scholar] [CrossRef]
- Yu, C.; He, S.; Zhu, W.; Ru, P.; Ge, X.; Govindasamy, K. Human Cytomegalovirus in Cancer: The Mechanism of HCMV-Induced Carcinogenesis and Its Therapeutic Potential. Front. Cell. Infect. Microbiol. 2023, 13, 1202138. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N. Viral Inhibition of MHC Class II Antigen Presentation. Trends Immunol. 2003, 24, 278–285. [Google Scholar] [CrossRef]
- Chang, W.L.W.; Barry, P.A. Attenuation of Innate Immunity by Cytomegalovirus IL-10 Establishes a Long-Term Deficit of Adaptive Antiviral Immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 22647–22652. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The Master Regulator of Immunity to Infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Del Prete, G.; De Carli, M.; Almerigogna, F.; Giudizi, M.G.; Biagiotti, R.; Romagnani, S. Human IL-10 Is Produced by Both Type 1 Helper (Th1) and Type 2 Helper (Th2) T Cell Clones and Inhibits Their Antigen-Specific Proliferation and Cytokine Production. J. Immunol. 1993, 150, 353–360. [Google Scholar] [CrossRef]
- Hynes, R.O. Alteration of Cell-Surface Proteins by Viral Transformation and by Proteolysis. Proc. Natl. Acad. Sci. USA 1973, 70, 3170–3174. [Google Scholar] [CrossRef]
- Raftery, M.J.; Wieland, D.; Gronewald, S.; Kraus, A.A.; Giese, T.; Schönrich, G. Shaping Phenotype, Function, and Survival of Dendritic Cells by Cytomegalovirus-Encoded IL-10. J. Immunol. 2004, 173, 3383–3391. [Google Scholar] [CrossRef] [PubMed]
- Cassatella, M.A.; Meda, L.; Bonora, S.; Ceska, M.; Constantin, G. Interleukin 10 (IL-10) Inhibits the Release of Proinflammatory Cytokines from Human Polymorphonuclear Leukocytes. Evidence for an Autocrine Role of Tumor Necrosis Factor and IL-1 Beta in Mediating the Production of IL-8 Triggered by Lipopolysaccharide. J. Exp. Med. 1993, 178, 2207–2211. [Google Scholar] [CrossRef] [PubMed]
- De Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; De Vries, J.E. Interleukin 10(IL-10) Inhibits Cytokine Synthesis by Human Monocytes: An Autoregulatory Role of IL-10 Produced by Monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef]
- Herbein, G. Tumors and Cytomegalovirus: An Intimate Interplay. Viruses 2022, 14, 812. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses Associated with Human Cancer. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 127–150. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Morales-Sánchez, A.; Fuentes-Pananá, E. Human Viruses and Cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef]
- Mui, U.; Haley, C.; Tyring, S. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. The Story of Human Cytomegalovirus and Cancer: Increasing Evidence and Open Questions. Neoplasia 2009, 11, 1–9. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- zur Hausen, H. The Search for Infectious Causes of Human Cancers: Where and Why (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5798–5808. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M. Malignant Transformation of Cells by Viruses. Perspect. Biol. Med. 1970, 14, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Pagano, J.S. Diseases and Mechanisms of Persistent DNA Virus Infection: Latency and Cellular Transformation. J. Infect. Dis. 1975, 132, 209–223. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Boldogh, I.; Huang, E.-S.; Baskar, J.F.; Gergely, L.; Albrecht, T. Oncogenic Transformation by Cellular DNA Isolated from Human Cytomegalovirus-Lnfected Cells. Intervirology 1992, 34, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G. Cellular Transformation by Human Cytomegalovirus. Cancers 2024, 16, 1970. [Google Scholar] [CrossRef]
- Abubakar, S.; Au, W.W.; Legator, M.S.; Albrecht, T. Induction of Chromosome Aberrations and Mitotic Arrest by Cytomegalovirus in Human Cells. Environ. Mutagen 1988, 12, 409–420. [Google Scholar] [CrossRef]
- Jault, F.M.; Jault, J.M.; Ruchti, F.; Fortunato, E.A.; Clark, C.; Corbeil, J.; Richman, D.D.; Spector, D.H. Cytomegalovirus Infection Induces High Levels of Cyclins, Phosphorylated Rb, and P53, Leading to Cell Cycle Arrest. J. Virol. 1995, 69, 6697–6704. [Google Scholar] [CrossRef]
- Ko, L.J.; Prives, C. P53: Puzzle and Paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef]
- VanDeusen, H.R.; Kalejta, R.F. The Retinoblastoma Tumor Suppressor Promotes Efficient Human Cytomegalovirus Lytic Replication. J. Virol. 2015, 89, 5012–5021. [Google Scholar] [CrossRef]
- Wang, J.; Marker, P.H.; Belcher, J.D.; Wilcken, D.E.L.; Burns, L.J.; Vercellotti, G.M.; Wang, X.L. Human Cytomegalovirus Immediate Early Proteins Upregulate Endothelial P53 Function. FEBS Lett. 2000, 474, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.; Weber, M.L.; Burns, L.J.; Jacob, H.S.; Vercellotti, G.M. Cytoplasmic Sequestration of P53 in Cytomegalovirus-Infected Human Endothelial Cells. Am. J. Pathol. 1996, 149, 1531–1539. [Google Scholar]
- Rosenke, K.; Samuel, M.A.; McDowell, E.T.; Toerne, M.A.; Fortunato, E.A. An Intact Sequence-Specific DNA-Binding Domain Is Required for Human Cytomegalovirus-Mediated Sequestration of P53 and May Promote in Vivo Binding to the Viral Genome during Infection. Virology 2006, 348, 19–34. [Google Scholar] [CrossRef]
- Wang, J.; Belcher, J.D.; Marker, P.H.; Wilcken, D.E.L.; Vercellotti, G.M.; Wang, X.L. Cytomegalovirus Inhibits P53 Nuclear Localization Signal Function. J. Mol. Med. 2001, 78, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; AbuBakar, S.; Albrecht, T. Activation of Proto-Oncogenes: An Immediate Early Event in Human Cytomegalovirus Infection. Science 1990, 247, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.P.; Yurochko, A.D.; Kowalik, T.F. Role of Human Cytomegalovirus Immediate-Early Proteins in Cell Growth Control. J. Virol. 2000, 74, 8028–8037. [Google Scholar] [CrossRef]
- Shen, Y.; Zhu, H.; Shenk, T. Human Cytomegalovirus IE1 and IE2 Proteins Are Mutagenic and Mediate “Hit-and-Run” Oncogenic Transformation in Cooperation with the Adenovirus E1A Proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 3341–3345. [Google Scholar] [CrossRef]
- Ouyang, P.; Rakus, K.; Van Beurden, S.J.; Westphal, A.H.; Davison, A.J.; Gatherer, D.; Vanderplasschen, A.F. IL-10 Encoded by Viruses: A Remarkable Example of Independent Acquisition of a Cellular Gene by Viruses and Its Subsequent Evolution in the Viral Genome. J. Gen. Virol. 2014, 95, 245–262. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Holmes, E.C. The Evolution of Large DNA Viruses: Combining Genomic Information of Viruses and Their Hosts. Trends Microbiol. 2004, 12, 458–465. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Chang, P.-C.; Wang, Y.; Li, M. Identification of Novel Viral Interleukin-10 Isoforms of Human Cytomegalovirus AD169. Virus Res. 2008, 131, 213–223. [Google Scholar] [CrossRef]
- Herbein, G. High-Risk Oncogenic Human Cytomegalovirus. Viruses 2022, 14, 2462. [Google Scholar] [CrossRef] [PubMed]
- Slobedman, B.; Barry, P.A.; Spencer, J.V.; Avdic, S.; Abendroth, A. Virus-Encoded Homologs of Cellular Interleukin-10 and Their Control of Host Immune Function. J. Virol. 2009, 83, 9618–9629. [Google Scholar] [CrossRef]
- Waters, S.; Lee, S.; Ariyanto, I.; Kresoje, N.; Leary, S.; Munyard, K.; Gaudieri, S.; Irish, A.; Keil, A.D.; Allcock, R.J.N.; et al. Sequencing of the Viral UL111a Gene Directly from Clinical Specimens Reveals Variants of HCMV-Encoded IL-10 That Are Associated with Altered Immune Responses to HCMV. Int. J. Mol. Sci. 2022, 23, 4644. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Garcia, W.; Abendroth, A.; Slobedman, B. Expression of a Human Cytomegalovirus Latency-Associated Homolog of Interleukin-10 during the Productive Phase of Infection. Virology 2008, 370, 285–294. [Google Scholar] [CrossRef]
- Pantaleão, S.Q.; Camillo, L.D.M.B.; Neves, T.C.; Menezes, I.D.G.; Stangherlin, L.M.; Batista, H.B.D.C.R.; Poole, E.; Nevels, M.; Philot, E.A.; Scott, A.L.; et al. Molecular Modelling of the HCMV IL-10 Protein Isoforms and Analysis of Their Interaction with the Human IL-10 Receptor. PLoS ONE 2022, 17, e0277953. [Google Scholar] [CrossRef]
- Sabat, R. IL-10 Family of Cytokines. Cytokine Growth Factor Rev. 2010, 21, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of Interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef]
- Avdic, S.; Cao, J.Z.; McSharry, B.P.; Clancy, L.E.; Brown, R.; Steain, M.; Gottlieb, D.J.; Abendroth, A.; Slobedman, B. Human Cytomegalovirus Interleukin-10 Polarizes Monocytes toward a Deactivated M2c Phenotype To Repress Host Immune Responses. J. Virol. 2013, 87, 10273–10282. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Gonçalves, R.M.-B.; Menier, C.; Dausset, J.; Carosella, E.D. Direct Evidence to Support the Role of HLA-G in Protecting the Fetus from Maternal Uterine Natural Killer Cytolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 11520–11525. [Google Scholar] [CrossRef]
- Charles, O.J.; Venturini, C.; Gantt, S.; Atkinson, C.; Griffiths, P.; Goldstein, R.A.; Breuer, J. Genomic and Geographical Structure of Human Cytomegalovirus. Proc. Natl. Acad. Sci. USA 2023, 120, e2221797120. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural Cytotoxic Activity of Peripheral-Blood Lymphocytes and Cancer Incidence: An 11-Year Follow-up Study of a General Population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Ruiz-García, R.; Vargas-Hernández, A.; Chinn, I.K.; Angelo, L.S.; Cao, T.N.; Coban-Akdemir, Z.; Jhangiani, S.N.; Meng, Q.; Forbes, L.R.; Muzny, D.M.; et al. Mutations in PI3K110δ Cause Impaired Natural Killer Cell Function Partially Rescued by Rapamycin Treatment. J. Allergy Clin. Immunol. 2018, 142, 605–617.e7. [Google Scholar] [CrossRef] [PubMed]
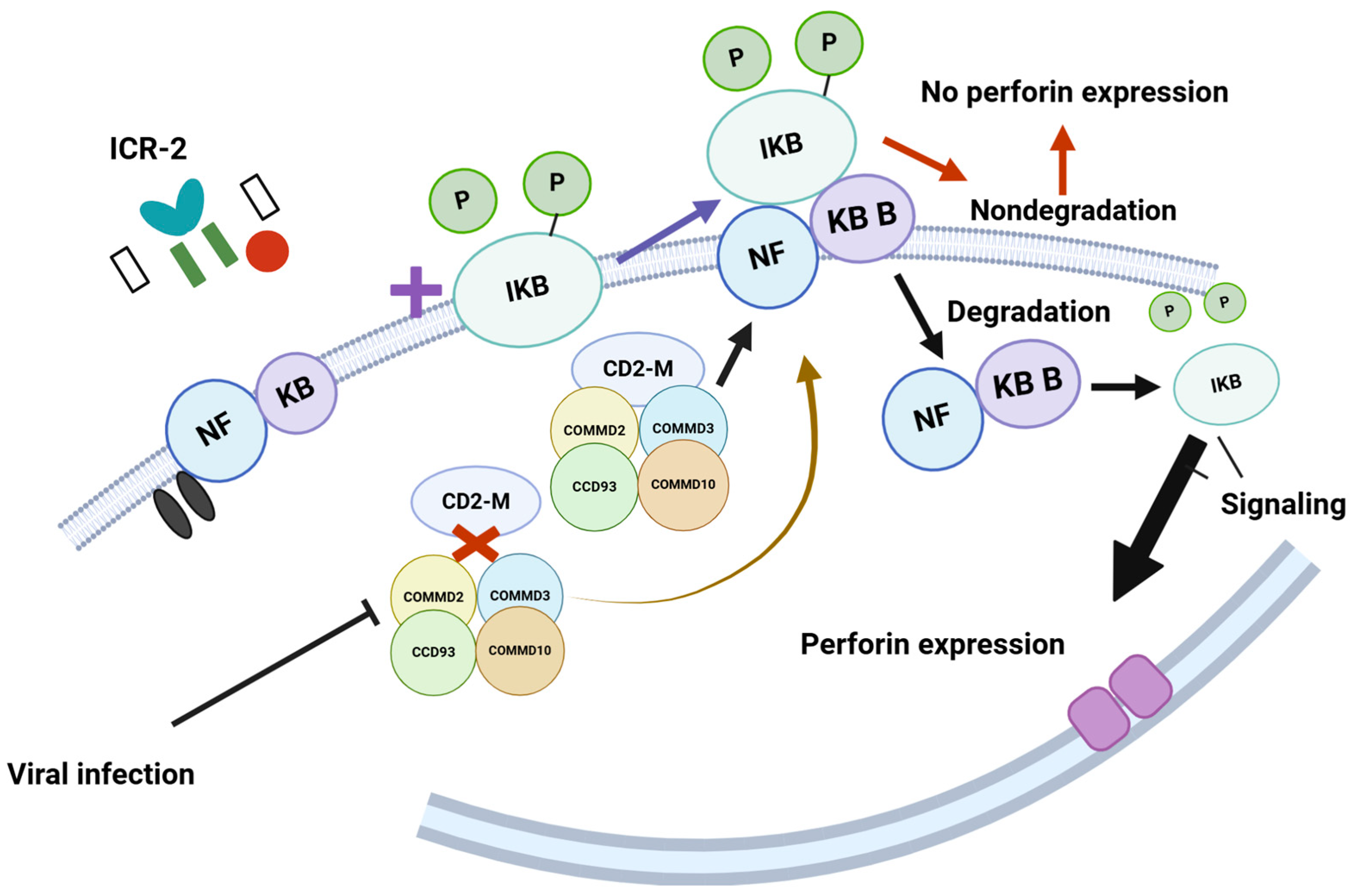
- Starokadomskyy, P.; Gluck, N.; Li, H.; Chen, B.; Wallis, M.; Maine, G.N.; Mao, X.; Zaidi, I.W.; Hein, M.Y.; McDonald, F.J.; et al. CCDC22 Deficiency in Humans Blunts Activation of Proinflammatory NF-κB Signaling. J. Clin. Investig. 2013, 123, 2244–2256. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Lichtenheld, M.G.; Meadows, G.G. A Role for NF-κB Activation in Perforin Expression of NK Cells Upon IL-2 Receptor Signaling. J. Immunol. 2002, 169, 1319–1325. [Google Scholar] [CrossRef]
- Yamashita, Y.; Nishikawa, A.; Iwahashi, Y.; Fujimoto, M.; Sasaki, I.; Mishima, H.; Kinoshita, A.; Hemmi, H.; Kanazawa, N.; Ohshima, K.; et al. Identification of a Novel CCDC22 Mutation in a Patient with Severe Epstein–Barr Virus-Associated Hemophagocytic Lymphohistiocytosis and Aggressive Natural Killer Cell Leukemia. Int. J. Hematol. 2019, 109, 744–750. [Google Scholar] [CrossRef]
- Moon, W.Y.; Powis, S.J. Does Natural Killer Cell Deficiency (NKD) Increase the Risk of Cancer? NKD May Increase the Risk of Some Virus Induced Cancer. Front. Immunol. 2019, 10, 1703. [Google Scholar] [CrossRef]
- Hughes, C.R.; Guasti, L.; Meimaridou, E.; Chuang, C.-H.; Schimenti, J.C.; King, P.J.; Costigan, C.; Clark, A.J.L.; Metherell, L.A. MCM4 Mutation Causes Adrenal Failure, Short Stature, and Natural Killer Cell Deficiency in Humans. J. Clin. Investig. 2012, 122, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S. Natural Killer Cell Deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; LeBlanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 Mutations Unify the Pathogenesis of Chronic Lymphoproliferative Disorders of NK Cells and T-Cell Large Granular Lymphocyte Leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Mahony, R.; Gargan, S.; Roberts, K.L.; Bourke, N.; Keating, S.E.; Bowie, A.G.; O’Farrelly, C.; Stevenson, N.J. A Novel Anti-Viral Role for STAT3 in IFN-α Signalling Responses. Cell. Mol. Life Sci. 2017, 74, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Ito, Y.; Kawabe, S.; Gotoh, K.; Takahashi, Y.; Kojima, S.; Naoe, T.; Esaki, S.; Kikuta, A.; Sawada, A.; et al. EBV-Associated T/NK–Cell Lymphoproliferative Diseases in Nonimmunocompromised Hosts: Prospective Analysis of 108 Cases. Blood 2012, 119, 673–686. [Google Scholar] [CrossRef]
- Lam, V.C.; Lanier, L.L. NK Cells in Host Responses to Viral Infections. Curr. Opin. Immunol. 2017, 44, 43–51. [Google Scholar] [CrossRef]
- Oh, J.H.; Kim, M.J.; Choi, S.J.; Ban, Y.H.; Lee, H.K.; Shin, E.-C.; Lee, K.-M.; Ha, S.-J. Sustained Type I Interferon Reinforces NK Cell–Mediated Cancer Immunosurveillance during Chronic Virus Infection. Cancer Immunol. Res. 2019, 7, 584–599. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein–Barr Virus in Chronic Active Infection and Haematological Malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef]
- Norris, P.J.; Hirschkorn, D.F.; DeVita, D.A.; Lee, T.-H.; Murphy, E.L. Human T Cell Leukemia Virus Type 1 Infection Drives Spontaneous Proliferation of Natural Killer Cells. Virulence 2010, 1, 19–28. [Google Scholar] [CrossRef]
- Gallant, R.E.; Arroyo, K.; Bracci, P.M.; Li, S.; Metayer, C.; Kogan, S.C.; Wendt, G.A.; Francis, S.S.; De Smith, A.J.; Wiemels, J.L. Clinical Characteristics of Cytomegalovirus-positive Pediatric Acute Lymphoblastic Leukemia at Diagnosis. Am. J. Hematol. 2022, 97, E198–E201. [Google Scholar] [CrossRef]
- Noyola, D.E.; Demmler, G.J.; Williamson, D.W.; Griesser, C.; Sellers, S.; Llorente, A.; Littman, T.; Williams, S.; Jarrett, L.; Yow, M.D. Cytomegalovirus Urinary Excretion and Long Term Outcome in Children with Congenital Cytomegalovirus Infection: Pediatr. Infect. Dis. J. 2000, 19, 505–510. [Google Scholar] [CrossRef]
- Francis, S.S.; Wallace, A.D.; Wendt, G.A.; Li, L.; Liu, F.; Riley, L.W.; Kogan, S.; Walsh, K.M.; De Smith, A.J.; Dahl, G.V.; et al. In Utero Cytomegalovirus Infection and Development of Childhood Acute Lymphoblastic Leukemia. Blood 2017, 129, 1680–1684. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Tasaki, M.; Saito, K.; Nakagawa, Y.; Ikeda, M.; Takahashi, K.; Tomita, Y. Long-Term CMV Monitoring and Chronic Rejection in Renal Transplant Recipients. Front. Cell. Infect. Microbiol. 2023, 13, 1190794. [Google Scholar] [CrossRef]
- Pastorczak, A.; Domka, K.; Fidyt, K.; Poprzeczko, M.; Firczuk, M. Mechanisms of Immune Evasion in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1536. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, G. Macrophages in Leukemia Microenvironment. Blood Sci. 2019, 1, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Derhovanessian, E.; Larbi, A. Immunosenescence and Cancer. Crit. Rev. Oncol. Hematol. 2010, 75, 165–172. [Google Scholar] [CrossRef]
- Takeuchi, M.; Miyoshi, H.; Ohshima, K. Tumor Microenvironment of Adult T-Cell Leukemia/Lymphoma. J. Clin. Exp. Hematop. 2021, 61, 202–209. [Google Scholar] [CrossRef]
- Green, M.L.; Leisenring, W.M.; Xie, H.; Walter, R.B.; Mielcarek, M.; Sandmaier, B.M.; Riddell, S.R.; Boeckh, M. CMV Reactivation after Allogeneic HCT and Relapse Risk: Evidence for Early Protection in Acute Myeloid Leukemia. Blood 2013, 122, 1316–1324. [Google Scholar] [CrossRef]
- Jang, J.E.; Hwang, D.Y.; Chung, H.; Kim, S.-J.; Eom, J.-I.; Jeung, H.-K.; Song, J.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. Early Cytomegalovirus Reactivation and Expansion of CD56brightCD16dim/−DNAM1+ Natural Killer Cells Are Associated with Antileukemia Effect after Haploidentical Stem Cell Transplantation in Acute Leukemia. Biol. Blood Marrow Transplant. 2019, 25, 2070–2078. [Google Scholar] [CrossRef]
- Manjappa, S.; Bhamidipati, P.K.; Stokerl-Goldstein, K.E.; DiPersio, J.F.; Uy, G.L.; Westervelt, P.; Liu, J.; Schroeder, M.A.; Vij, R.; Abboud, C.N.; et al. Protective Effect of Cytomegalovirus Reactivation on Relapse after Allogeneic Hematopoietic Cell Transplantation in Acute Myeloid Leukemia Patients Is Influenced by Conditioning Regimen. Biol. Blood Marrow Transplant. 2014, 20, 46–52. [Google Scholar] [CrossRef]
- Bigley, A.B.; Baker, F.L.; Simpson, R.J. Cytomegalovirus: An Unlikely Ally in the Fight against Blood Cancers? Clin. Exp. Immunol. 2018, 193, 265–274. [Google Scholar] [CrossRef]
- Samudio, I.; Rezvani, K.; Shaim, H.; Hofs, E.; Ngom, M.; Bu, L.; Liu, G.; Lee, J.T.C.; Imren, S.; Lam, V.; et al. UV-Inactivated HSV-1 Potently Activates NK Cell Killing of Leukemic Cells. Blood 2016, 127, 2575–2586. [Google Scholar] [CrossRef]
- Müller, L.M.E.; Holmes, M.; Michael, J.L.; Scott, G.B.; West, E.J.; Scott, K.J.; Parrish, C.; Hall, K.; Stäble, S.; Jennings, V.A.; et al. Plasmacytoid Dendritic Cells Orchestrate Innate and Adaptive Anti-Tumor Immunity Induced by Oncolytic Coxsackievirus A21. J. Immunother. Cancer 2019, 7, 164. [Google Scholar] [CrossRef]
- Pupuleku, A.; Costa-García, M.; Farré, D.; Hengel, H.; Angulo, A.; Muntasell, A.; López-Botet, M. Elusive Role of the CD94/NKG2C NK Cell Receptor in the Response to Cytomegalovirus: Novel Experimental Observations in a Reporter Cell System. Front. Immunol. 2017, 8, 1317. [Google Scholar] [CrossRef] [PubMed]
- Gumá, M.; Angulo, A.; Vilches, C.; Gómez-Lozano, N.; Malats, N.; López-Botet, M. Imprint of Human Cytomegalovirus Infection on the NK Cell Receptor Repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Kreutzman, A.; Ladell, K.; Koechel, C.; Gostick, E.; Ekblom, M.; Stenke, L.; Melo, T.; Einsele, H.; Porkka, K.; Price, D.A.; et al. Expansion of Highly Differentiated CD8+ T-Cells or NK-Cells in Patients Treated with Dasatinib Is Associated with Cytomegalovirus Reactivation. Leukemia 2011, 25, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, K.; Kitawaki, T.; Sugimoto, N.; Sozu, T.; Anzai, N.; Okada, M.; Nohgawa, M.; Hatanaka, K.; Arima, N.; Ishikawa, T.; et al. Principal Component Analysis Uncovers Cytomegalovirus-Associated NK Cell Activation in Ph+ Leukemia Patients Treated with Dasatinib. Leukemia 2017, 31, 203–212. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Falco, M.; Bertaina, A.; Muccio, L.; Alicata, C.; Frassoni, F.; Locatelli, F.; Moretta, L.; Moretta, A. Human Cytomegalovirus Infection Promotes Rapid Maturation of NK Cells Expressing Activating Killer Ig–like Receptor in Patients Transplanted with NKG2C−/− Umbilical Cord Blood. J. Immunol. 2014, 192, 1471–1479. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTEIN | IMMUNE EVASION MECHANISMS | REFERENCES |
| gB | Interaction with cell integrins. Tropism and the Facilitation of Complete Infection. Entry into fibroblasts and epithelial cells. Formation of a pentameric complex with gH and gL; improves viral entry in epithelial, endothelial, monocytic and dendritic cells, which relates to infection levels. The processes of viral cell attachment and replication. Retention of MHC class I and reduction in MHC class II expression. Promotion of the degradation of MHC class I. Promotion of MHC I degradation. Retention of MHC class I heavy chains and the induction of HLA-G degradation. Inhibition of TAP and the process of peptide translocation. MHC Class I proteins experience a delay when transporting from the endoplasmic reticulum (ER) to the Golgi apparatus. Prevention of the formation of viral antigenic peptides and the response to interferons. | [21] |
| gH/gL | [21] | |
| gO | [21] | |
| UL128, UL130, and UL131a | [21] | |
| gM/gN | [21] | |
| US3 | [22,23,24,25] | |
| US11 | [26,27] | |
| US2 | [28,29] | |
| US10 | [30,31] | |
| US6 | [32,33] | |
| pp71 | [34] | |
| pp65 | [35] |
| TOWNE | TOLEDO | AD169 | MERLIN | 56.1 | 57.1 | 58.1 | 59.1 | 60.1 | 61.1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| hIL-10 | 42.75 | 41.28 | 40.70 | 41.44 | 46.0 | 51.17 | 49.60 | 48.54 | 50.30 | 49.14 |
| TOWNE | TOLEDO | AD169 | MERLIN | 56.1 | 57.1 | 58.1 | 10 59.1 | 10 60.1 | 61.1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| TOWNE | 100 | 89.39 | 83.09 | 83.48 | 87.07 | 68.47 | 78.44 | 59.01 | 62.21 | 68.98 |
| TOLEDO | 100 | 83.03 | 82.87 | 86.31 | 67.31 | 77.30 | 57.98 | 61.05 | 67.95 | |
| AD169 | 100 | 82.99 | 86.44 | 67.57 | 77.30 | 58.24 | 61.18 | 67.95 | ||
| MERLIN | 100 | 87.07 | 68.48 | 78.06 | 58.72 | 61.70 | 70.08 | |||
| 56.1 | 100 | 73.17 | 83.62 | 63.36 | 66.58 | 73.44 | ||||
| 57.1 | 100 | 84.33 | 80.95 | 89.15 | 77.30 | |||||
| 58.1 | 100 | 70.34 | 76.84 | 82.12 | ||||||
| 59.1 | 100 | 77.18 | 82.59 | |||||||
| 60.1 | 100 | 72.87 | ||||||||
| 61.1 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pamela, R.-H.; Minerva, M.-R.; Ernesto, C.-M.M.; Manuel, M.-A.J.; Norberto, S.-E.; Francisco, A.-H.; de la Torre Silvia, M.-D.; Angélica, R.-L.; Elva, J.-H.; Carlos, N.-E.J.; et al. Is the vIL-10 Protein from Cytomegalovirus Associated with the Potential Development of Acute Lymphoblastic Leukemia? Viruses 2025, 17, 435. https://doi.org/10.3390/v17030435
Pamela R-H, Minerva M-R, Ernesto C-MM, Manuel M-AJ, Norberto S-E, Francisco A-H, de la Torre Silvia M-D, Angélica R-L, Elva J-H, Carlos N-EJ, et al. Is the vIL-10 Protein from Cytomegalovirus Associated with the Potential Development of Acute Lymphoblastic Leukemia? Viruses. 2025; 17(3):435. https://doi.org/10.3390/v17030435
Chicago/Turabian StylePamela, Ruvalcaba-Hernández, Mata-Rocha Minerva, Cruz-Muñoz Mario Ernesto, Mejía-Aranguré Juan Manuel, Sánchez-Escobar Norberto, Arenas-Huertero Francisco, Melchor-Doncel de la Torre Silvia, Rangel-López Angélica, Jiménez-Hernández Elva, Nuñez-Enriquez Juan Carlos, and et al. 2025. "Is the vIL-10 Protein from Cytomegalovirus Associated with the Potential Development of Acute Lymphoblastic Leukemia?" Viruses 17, no. 3: 435. https://doi.org/10.3390/v17030435
APA StylePamela, R.-H., Minerva, M.-R., Ernesto, C.-M. M., Manuel, M.-A. J., Norberto, S.-E., Francisco, A.-H., de la Torre Silvia, M.-D., Angélica, R.-L., Elva, J.-H., Carlos, N.-E. J., Sara, O., Juan, X.-C., Ariadnna, C.-C., Paula, F.-A., & José, A.-G. (2025). Is the vIL-10 Protein from Cytomegalovirus Associated with the Potential Development of Acute Lymphoblastic Leukemia? Viruses, 17(3), 435. https://doi.org/10.3390/v17030435