
APOBEC3 Proteins: From Antiviral Immunity to Oncogenic Drivers in HPV-Positive Cancers
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
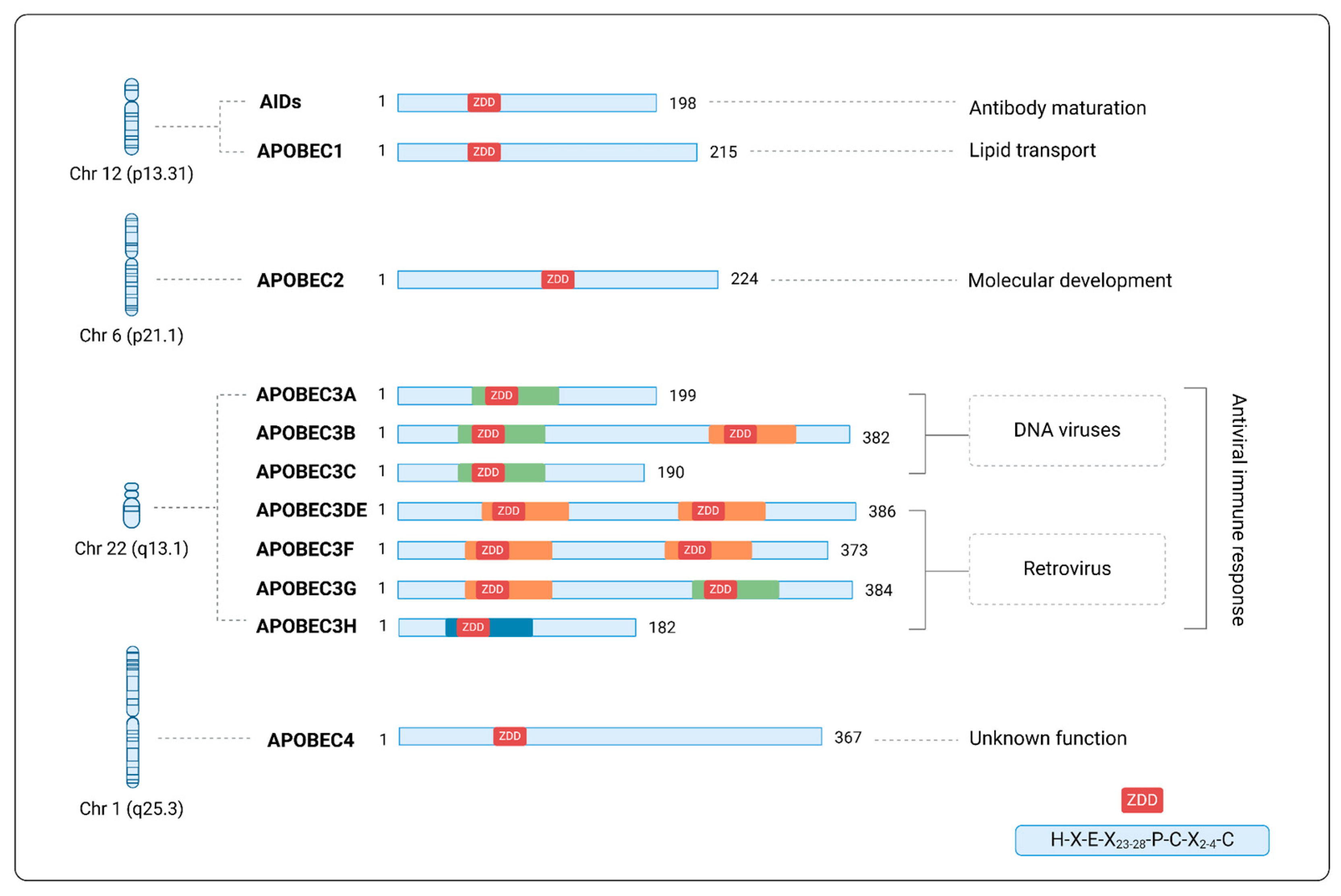
2. APOBEC Superfamily
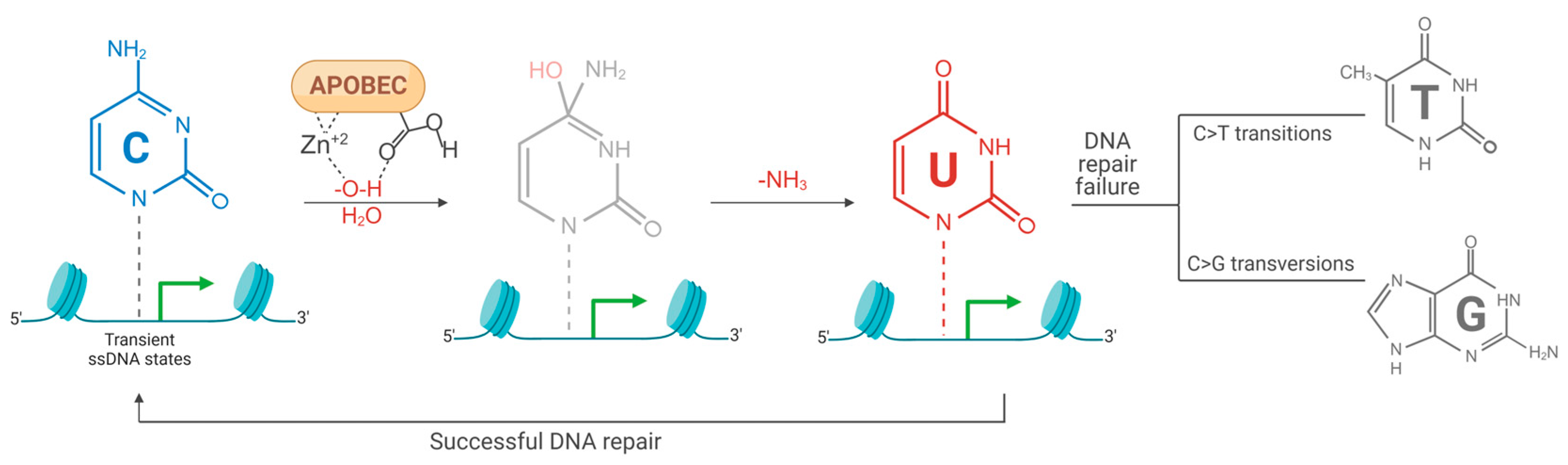
3. Antiviral Activity of APOBEC3 Proteins
4. Hypermutation in the Cellular Genome
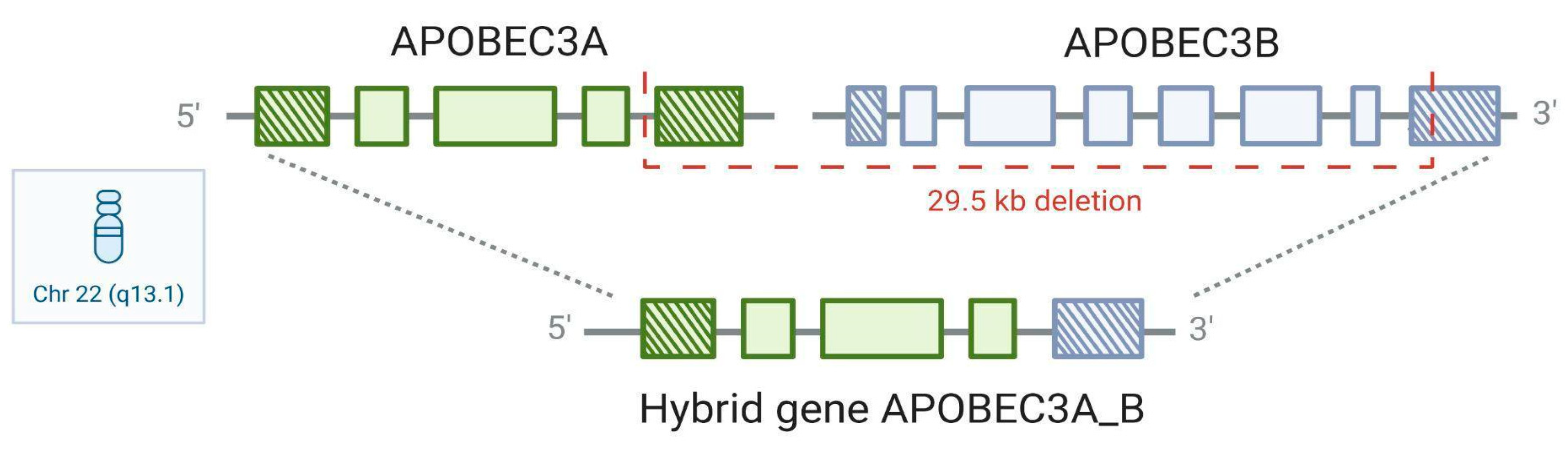
5. APOBEC3A_B Polymorphism
6. APOBEC3A_B Deletion Polymorphism Frequency, Population Variation, and Cancer Risk
7. HPV-Related Cancers and APOBEC3A_B Polymorphism
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mertz, T.M.; Collins, C.D.; Dennis, M.; Coxon, M.; Roberts, S.A. APOBEC-Induced Mutagenesis in Cancer. Annu. Rev. Genet. 2022, 56, 229–252. [Google Scholar] [CrossRef] [PubMed]
- Pecori, R.; Di Giorgio, S.; Paulo Lorenzo, J.; Nina Papavasiliou, F. Functions and Consequences of AID/APOBEC-Mediated DNA and RNA Deamination. Nat. Rev. Genet. 2022, 23, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Dananberg, A.; Striepen, J.; Rozowsky, J.S.; Petljak, M. APOBEC Mutagenesis in Cancer Development and Susceptibility. Cancers 2024, 16, 374. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.J.; Fenton, T.R. The APOBEC3 Genes and Their Role in Cancer: Insights from Human Papillomavirus. J. Mol. Endocrinol. 2019, 62, R269–R287. [Google Scholar] [CrossRef]
- Hata, A.N.; Larijani, M. Targeting APOBECs in Cancer: It’s about Timing. Cancer Cell 2024, 42, 497–501. [Google Scholar] [CrossRef]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A Can Activate the DNA Damage Response and Cause Cell-Cycle Arrest. EMBO Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef]
- Chervova, A.; Fatykhov, B.; Koblov, A.; Shvarov, E.; Preobrazhenskaya, J.; Vinogradov, D.; Ponomarev, G.V.; Gelfand, M.S.; Kazanov, M.D. Analysis of Gene Expression and Mutation Data Points on Contribution of Transcription to the Mutagenesis by APOBEC Enzymes. NAR Cancer 2021, 3, zcab025. [Google Scholar] [CrossRef]
- Castilha, E.P.; Curti, R.R.d.J.; de Oliveira, J.N.; Vitiello, G.A.F.; Guembarovski, R.L.; Couto-Filho, J.d.; Oliveira, K.B.d. APOBEC3A/B Polymorphism Is Not Associated with Human Papillomavirus Infection and Cervical Carcinogenesis. Pathogens 2023, 12, 636. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a Germline Copy Number Polymorphism of APOBEC3A and APOBEC3B with Burden of Putative APOBEC-Dependent Mutations in Breast Cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Guo, K.; Liu, C.-W.; Santiago, M.L.; Pyeon, D. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J. Virol. 2018, 92, 1–17. [Google Scholar] [CrossRef]
- Warren, C.J.; Santiago, M.L.; Pyeon, D. APOBEC3: Friend or Foe in Human Papillomavirus Infection and Oncogenesis? Annu. Rev. Virol. 2022, 9, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Weitzman, M.D. The Spectrum of APOBEC3 Activity: From Anti-Viral Agents to Anti-Cancer Opportunities. DNA Repair 2019, 83, 102700. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiu, X.; Zhang, N.; Wang, Y.; Wang, M.; Li, D.; Wang, L.; Du, Y. APOBEC-Mediated Genomic Alterations Link Immunity and Viral Infection during Human Papillomavirus-Driven Cervical Carcinogenesis. Biosci. Trends 2017, 11, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Fenton, T. APOBEC3 Genes: Retroviral Restriction Factors to Cancer Drivers. Trends Mol. Med. 2015, 21, 274–284. [Google Scholar] [CrossRef]
- Revathidevi, S.; Murugan, A.K.; Nakaoka, H.; Inoue, I.; Munirajan, A.K. APOBEC: A Molecular Driver in Cervical Cancer Pathogenesis. Cancer Lett. 2021, 496, 104–116. [Google Scholar] [CrossRef]
- Riva, G.; Albano, C.; Gugliesi, F.; Pasquero, S.; Pacheco, S.F.C.; Pecorari, G.; Landolfo, S.; Biolatti, M.; Dell’Oste, V. HPV Meets APOBEC: New Players in Head and Neck Cancer. Int. J. Mol. Sci. 2021, 22, 1402. [Google Scholar] [CrossRef]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. In Trends in Biochemical Sciences; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; pp. 578–594. [Google Scholar] [CrossRef]
- Cervantes-Gracia, K.; Gramalla-Schmitz, A.; Weischedel, J.; Chahwan, R. APOBECs Orchestrate Genomic and Epigenomic Editing across Health and Disease. Trends Genet. 2021, 37, 1028–1043. [Google Scholar] [CrossRef]
- Warren, C.; Westrich, J.; Doorslaer, K.; Pyeon, D. Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses 2017, 9, 233. [Google Scholar] [CrossRef]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC Deaminases and Cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef]
- Lo, C.C.; Coschigano, K.T. ApoB48 as an Efficient Regulator of Intestinal Lipid Transport. Front. Physiol. 2020, 11, 796. [Google Scholar] [CrossRef]
- Nakajima, K.; Nagamine, T.; Fujita, M.Q.; Ai, M.; Tanaka, A.; Schaefer, E. Apolipoprotein B-48: A Unique Marker of Chylomicron Metabolism. In Advances in Clinical Chemistry; Academic Press Inc.: Cambridge, MA, USA, 2014; Volume 64, pp. 117–177. [Google Scholar] [CrossRef]
- Vieira, V.C.; Soares, M.A. The Role of Cytidine Deaminases on Innate Immune Responses against Human Viral Infections. BioMed Res. Int. 2013, 2013, 1–18. [Google Scholar] [CrossRef] [PubMed]
- McCool, M.A.; Bryant, C.J.; Abriola, L.; Surovtseva, Y.V.; Baserga, S.J. The Cytidine Deaminase APOBEC3A Regulates Nucleolar Function to Promote Cell Growth and Ribosome Biogenesis. PLoS Biol. 2024, 22, e3002718. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Kosugi, Y.; Ito, J.; Sato, K. The Battle between Retroviruses and APOBEC3 Genes: Its Past and Present. Viruses 2021, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.M.; Sudarsan, A.K.A.; Shayeganmehr, A.; Munhoz, E.P.; Lao, R.; Gaba, A.; Rodríguez, M.G.; Love, R.P.; Polacco, B.J.; Zhou, Y.; et al. Protein Interaction Map of APOBEC3 Enzyme Family Reveals Deamination-Independent Role in Cellular Function. Mol. Cell. Proteom. 2024, 23, 100755. [Google Scholar] [CrossRef]
- Kidd, J.M.; Newman, T.L.; Tuzun, E.; Kaul, R.; Eichler, E.E. Population Stratification of a Common APOBEC Gene Deletion Polymorphism. PLoS Genet. 2007, 3, e63. [Google Scholar] [CrossRef]
- Jha, P.; Sinha, S.; Kanchan, K.; Qidwai, T.; Narang, A.; Singh, P.K.; Pati, S.S.; Mohanty, S.; Mishra, S.K.; Sharma, S.K.; et al. Deletion of the APOBEC3B Gene Strongly Impacts Susceptibility to Falciparum Malaria. Infect. Genet. Evol. 2012, 12, 142–148. [Google Scholar] [CrossRef]
- Sadeghpour, S.; Khodaee, S.; Rahnama, M.; Rahimi, H.; Ebrahimi, D. Human APOBEC3 Variations and Viral Infection. Viruses 2021, 13, 1366. [Google Scholar] [CrossRef]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A Functions as a Restriction Factor of Human Papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad Antiretroviral Defence by Human APOBEC3G through Lethal Editing of Nascent Reverse Transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Delviks-Frankenberry, K.A.; Nikolaitchik, O.A.; Burdick, R.C.; Gorelick, R.J.; Keele, B.F.; Hu, W.-S.; Pathak, V.K. Minimal Contribution of APOBEC3-Induced G-to-A Hypermutation to HIV-1 Recombination and Genetic Variation. PLoS Pathog. 2016, 12, e1005646. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takeuchi, J.S.; Misawa, N.; Izumi, T.; Kobayashi, T.; Kimura, Y.; Iwami, S.; Takaori-Kondo, A.; Hu, W.-S.; Aihara, K.; et al. APOBEC3D and APOBEC3F Potently Promote HIV-1 Diversification and Evolution in Humanized Mouse Model. PLoS Pathog. 2014, 10, e1004453. [Google Scholar] [CrossRef]
- Yu, Q.; Chen, D.; König, R.; Mariani, R.; Unutmaz, D.; Landau, N.R. APOBEC3B and APOBEC3C Are Potent Inhibitors of Simian Immunodeficiency Virus Replication. J. Biol. Chem. 2004, 279, 53379–53386. [Google Scholar] [CrossRef]
- Thielen, B.K.; McNevin, J.P.; McElrath, M.J.; Hunt, B.V.S.; Klein, K.C.; Lingappa, J.R. Innate Immune Signaling Induces High Levels of TC-Specific Deaminase Activity in Primary Monocyte-Derived Cells through Expression of APOBEC3A Isoforms. J. Biol. Chem. 2010, 285, 27753–27766. [Google Scholar] [CrossRef]
- Kim, K.; Calabrese, P.; Wang, S.; Qin, C.; Rao, Y.; Feng, P.; Chen, X.S. The Roles of APOBEC-Mediated RNA Editing in SARS-CoV-2 Mutations, Replication and Fitness. Sci. Rep. 2022, 12, 14972. [Google Scholar] [CrossRef]
- Janahi, E.M.; McGarvey, M.J. The Inhibition of Hepatitis B Virus by APOBEC Cytidine Deaminases. J. Viral Hepatitis 2013, 20, 821–828. [Google Scholar] [CrossRef]
- Luo, X.; Huang, Y.; Chen, Y.; Tu, Z.; Hu, J.; Tavis, J.E.; Huang, A.; Hu, Y. Association of Hepatitis B Virus Covalently Closed Circular DNA and Human APOBEC3B in Hepatitis B Virus-Related Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0157708. [Google Scholar] [CrossRef]
- Lehle, J.; Soleimanpour, M.; Mokhtari, S.; Ebrahimi, D. Viral Infection, APOBEC3 Dysregulation, and Cancer. Front. Genet. 2024, 15, 1489324. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Moraes, S.N.; Shaban, N.M.; Fanunza, E.; Bierle, C.J.; Southern, P.J.; Bresnahan, W.A.; Rice, S.A.; Harris, R.S. APOBECs and Herpesviruses. Viruses 2021, 13, 390. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Cao, Y.; Yang, Z. Roles of APOBEC3 in Hepatitis B Virus (HBV) Infection and Hepatocarcinogenesis. Bioengineered 2021, 12, 2074–2086. [Google Scholar] [CrossRef] [PubMed]
- Wakae, K.; Kondo, S.; Pham, H.T.; Wakisaka, N.; Que, L.; Li, Y.; Zheng, X.; Fukano, K.; Kitamura, K.; Watashi, K.; et al. EBV-LMP1 Induces APOBEC3s and Mitochondrial DNA Hypermutation in Nasopharyngeal Cancer. Cancer Med. 2020, 9, 7663–7671. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.-P.; Guétard, D.; Henry, M.; Wain-Hobson, S. Evidence for Editing of Human Papillomavirus DNA by APOBEC3 in Benign and Precancerous Lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef]
- Hirose, Y.; Yamaguchi-Naka, M.; Onuki, M.; Tenjimbayashi, Y.; Tasaka, N.; Satoh, T.; Tanaka, K.; Iwata, T.; Sekizawa, A.; Matsumoto, K.; et al. High Levels of Within-Host Variations of Human Papillomavirus 16 E1/E2 Genes in Invasive Cervical Cancer. Front. Microbiol. 2020, 11, 596334. [Google Scholar] [CrossRef]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 Deaminases Induce Hypermutation in Human Papillomavirus 16 DNA upon Beta Interferon Stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef]
- Della Fera, A.N.; Warburton, A.; Coursey, T.L.; Khurana, S.; McBride, A.A. Persistent Human Papillomavirus Infection. Viruses 2021, 13, 321. [Google Scholar] [CrossRef]
- Wallace, N.A.; Münger, K. The Curious Case of APOBEC3 Activation by Cancer-Associated Human Papillomaviruses. PLoS Pathog. 2018, 14, e1006717. [Google Scholar] [CrossRef]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T.; et al. Mutations in the HPV16 Genome Induced by APOBEC3 Are Associated with Viral Clearance. Nat. Commun. 2020, 11, 886. [Google Scholar] [CrossRef]
- Warren, C.J.; Van Doorslaer, K.; Pandey, A.; Espinosa, J.M.; Pyeon, D. Role of the Host Restriction Factor APOBEC3 on Papillomavirus Evolution. Virus Evol. 2015, 1, vev015. [Google Scholar] [CrossRef]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Luengas, E.M.; McCann, J.L.; Ebrahimi, D.; Temiz, N.A.; Love, R.P.; Feng, Y.; Adolph, M.B.; Chelico, L.; Law, E.K.; et al. The DNA Cytosine Deaminase APOBEC3H Haplotype I Likely Contributes to Breast and Lung Cancer Mutagenesis. Nat. Commun. 2016, 7, 12918. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sun, D.S.; Yoon, J.; Ko, Y.H.; Won, H.S.; Kim, J.S. Clinical Implications of APOBEC3A and 3B Expression in Patients with Breast Cancer. PLoS ONE 2020, 15, e0230261. [Google Scholar] [CrossRef] [PubMed]
- Cannataro, V.L.; Gaffney, S.G.; Sasaki, T.; Issaeva, N.; Grewal, N.K.S.; Grandis, J.R.; Yarbrough, W.G.; Burtness, B.; Anderson, K.S.; Townsend, J.P. APOBEC-Induced Mutations and Their Cancer Effect Size in Head and Neck Squamous Cell Carcinoma. Oncogene 2019, 38, 3475–3487. [Google Scholar] [CrossRef]
- Argyris, P.P.; Wilkinson, P.E.; Jarvis, M.C.; Magliocca, K.R.; Patel, M.R.; Vogel, R.I.; Gopalakrishnan, R.; Koutlas, I.G.; Harris, R.S. Endogenous APOBEC3B Overexpression Characterizes HPV-Positive and HPV-Negative Oral Epithelial Dysplasias and Head and Neck Cancers. Mod. Pathol. 2021, 34, 280–290. [Google Scholar] [CrossRef]
- Chen, T.-W.; Lee, C.-C.; Liu, H.; Wu, C.-S.; Pickering, C.R.; Huang, P.-J.; Wang, J.; Chang, I.Y.-F.; Yeh, Y.-M.; Chen, C.-D.; et al. APOBEC3A Is an Oral Cancer Prognostic Biomarker in Taiwanese Carriers of an APOBEC Deletion Polymorphism. Nat. Commun. 2017, 8, 465. [Google Scholar] [CrossRef]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B.; et al. Mechanisms of APOBEC3 Mutagenesis in Human Cancer Cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B Mutagenesis in Multiple Human Cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef]
- Chan, K.; Roberts, S.A.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.J.; Fargo, D.C.; Mieczkowski, P.A.; et al. An APOBEC3A Hypermutation Signature Is Distinguishable from the Signature of Background Mutagenesis by APOBEC3B in Human Cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef]
- Jalili, P.; Bowen, D.; Langenbucher, A.; Park, S.; Aguirre, K.; Corcoran, R.B.; Fleischman, A.G.; Lawrence, M.S.; Zou, L.; Buisson, R. Quantification of Ongoing APOBEC3A Activity in Tumor Cells by Monitoring RNA Editing at Hotspots. Nat. Commun. 2020, 11, 2971. [Google Scholar] [CrossRef]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-Mediated Cytosine Deamination Links PIK3CA Helical Domain Mutations to Human Papillomavirus-Driven Tumor Development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.-F.; Shandilya, S.M.D.; Silvas, T.V.; Nalivaika, E.A.; Kouno, T.; Kelch, B.A.; Ryder, S.P.; Kurt-Yilmaz, N.; Somasundaran, M.; Schiffer, C.A. The SsDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure 2015, 23, 903–911. [Google Scholar] [CrossRef]
- Klonowska, K.; Kluzniak, W.; Rusak, B.; Jakubowska, A.; Ratajska, M.; Krawczynska, N.; Vasilevska, D.; Czubak, K.; Wojciechowska, M.; Cybulski, C.; et al. The 30 Kb Deletion in the APOBEC3 Cluster Decreases APOBEC3A and APOBEC3B Expression and Creates a Transcriptionally Active Hybrid Gene but Does Not Associate with Breast Cancer in the European Population. Oncotarget 2017, 8, 76357–76374. [Google Scholar] [CrossRef]
- Caval, V.; Suspène, R.; Shapira, M.; Vartanian, J.-P.; Wain-Hobson, S. A Prevalent Cancer Susceptibility APOBEC3A Hybrid Allele Bearing APOBEC3B 3′UTR Enhances Chromosomal DNA Damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef]
- Revathidevi, S.; Manikandan, M.; Rao, A.K.D.M.; Vinothkumar, V.; Arunkumar, G.; Rajkumar, K.S.; Ramani, R.; Rajaraman, R.; Ajay, C.; Munirajan, A.K. Analysis of APOBEC3A/3B Germline Deletion Polymorphism in Breast, Cervical and Oral Cancers from South India and Its Impact on MiRNA Regulation. Tumor Biol. 2016, 37, 11983–11990. [Google Scholar] [CrossRef]
- Vitiello, G.A.F.; Amarante, M.K.; Banin-Hirata, B.K.; Campos, C.Z.; de Oliveira, K.B.; Losi-Guembarovski, R.; Watanabe, M.A.E. Transforming Growth Factor Beta Receptor II (TGFBR2) Promoter Region Polymorphism in Brazilian Breast Cancer Patients: Association with Susceptibility, Clinicopathological Features, and Interaction with TGFB1 Haplotypes. Breast Cancer Res. Treat. 2019, 178, 207–219. [Google Scholar] [CrossRef]
- Gansmo, L.B.; Romundstad, P.; Hveem, K.; Vatten, L.; Nik-Zainal, S.; Lønning, P.E.; Knappskog, S. APOBEC3A/B Deletion Polymorphism and Cancer Risk. Carcinogenesis 2018, 39, 118–124. [Google Scholar] [CrossRef]
- Gansmo, L.B.; Sofiyeva, N.; Bjørnslett, M.; Romundstad, P.; Hveem, K.; Vatten, L.; Dørum, A.; Lønning, P.E.; Knappskog, S. Impact of the APOBEC3A/B Deletion Polymorphism on Risk of Ovarian Cancer. Sci. Rep. 2021, 11, 23463. [Google Scholar] [CrossRef]
- Long, J.; Delahanty, R.J.; Li, G.; Gao, Y.-T.; Lu, W.; Cai, Q.; Xiang, Y.-B.; Li, C.; Ji, B.-T.; Zheng, Y.; et al. A Common Deletion in the APOBEC3 Genes and Breast Cancer Risk. JNCI J. Natl. Cancer Inst. 2013, 105, 573–579. [Google Scholar] [CrossRef]
- Rezaei, M.; Hashemi, M.; Hashemi, S.M.; Mashhadi, M.A.; Taheri, M. APOBEC3 Deletion Is Associated with Breast Cancer Risk in a Sample of Southeast Iranian Population. Int. J. Mol. Cell Med. 2015, 4, 103–108. [Google Scholar]
- Wen, W.X.; Soo, J.S.-S.; Kwan, P.Y.; Hong, E.; Khang, T.F.; Mariapun, S.; Lee, C.S.-M.; Hasan, S.N.; Rajadurai, P.; Yip, C.H.; et al. Germline APOBEC3B Deletion Is Associated with Breast Cancer Risk in an Asian Multi-Ethnic Cohort and with Immune Cell Presentation. Breast Cancer Res. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Xuan, D.; Li, G.; Cai, Q.; Deming-Halverson, S.; Shrubsole, M.J.; Shu, X.-O.; Kelley, M.C.; Zheng, W.; Long, J. APOBEC3 Deletion Polymorphism Is Associated with Breast Cancer Risk among Women of European Ancestry. Carcinogenesis 2013, 34, 2240–2243. [Google Scholar] [CrossRef] [PubMed]
- Göhler, S.; Da Silva Filho, M.I.; Johansson, R.; Enquist-Olsson, K.; Henriksson, R.; Hemminki, K.; Lenner, P.; Försti, A. Impact of Functional Germline Variants and a Deletion Polymorphism in APOBEC3A and APOBEC3B on Breast Cancer Risk and Survival in a Swedish Study Population. J. Cancer Res. Clin. Oncol. 2016, 142, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Marouf, C.; Göhler, S.; Filho, M.I.D.S.; Hajji, O.; Hemminki, K.; Nadifi, S.; Försti, A. Analysis of Functional Germline Variants in APOBEC3 and Driver Genes on Breast Cancer Risk in Moroccan Study Population. BMC Cancer 2016, 16, 165. [Google Scholar] [CrossRef]
- Hashemi, M.; Moazeni-Roodi, A.; Taheri, M. Association of APOBEC3 Deletion with Cancer Risk: A Meta-analysis of 26 225 Cases and 37 201 Controls. Asia Pac. J. Clin. Oncol. 2019, 15, 275–287. [Google Scholar] [CrossRef]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining “Mutation” and “Polymorphism” in the Era of Personal Genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC Cytidine Deaminase Mutagenesis Pattern Is Widespread in Human Cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of Genomic Alterations in Cervical Carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Burk, R.D.; Chen, Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated Genomic and Molecular Characterization of Cervical Cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Feber, A.; Worth, D.C.; Chakravarthy, A.; de Winter, P.; Shah, K.; Arya, M.; Saqib, M.; Nigam, R.; Malone, P.R.; Tan, W.S.; et al. CSN1 Somatic Mutations in Penile Squamous Cell Carcinoma. Cancer Res. 2016, 76, 4720–4727. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Gan, J.; Feng, X.; Zhang, M.; Chen, Z.; Zhao, H.; Du, Y. APOBEC3B Is Overexpressed in Cervical Cancer and Promotes the Proliferation of Cervical Cancer Cells through Apoptosis, Cell Cycle, and P53 Pathway. Front. Oncol. 2022, 12, 864889. [Google Scholar] [CrossRef]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human Papillomavirus E6 Triggers Upregulation of the Antiviral and Cancer Genomic DNA Deaminase APOBEC3B. mBio 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Butt, Y.; Sakhtemani, R.; Mohamad-Ramshan, R.; Lawrence, M.S.; Bhagwat, A.S. Distinguishing Preferences of Human APOBEC3A and APOBEC3B for Cytosines in Hairpin Loops, and Reflection of These Preferences in APOBEC-Signature Cancer Genome Mutations. Nat. Commun. 2024, 15, 2369. [Google Scholar] [CrossRef]
- Pan, J.; Zabidi, M.M.A.; Chong, B.; Meng, M.; Ng, P.; Hasan, S.N.; Sandey, B.; Bahnu, S.; Rajadurai, P.; Yip, C.; et al. Germline APOBEC3B Deletion Increases Somatic Hypermutation in Asian Breast Cancer That Is Associated with Her2 Subtype, PIK3CA Mutations and Immune Activation. Int. J. Cancer 2021, 148, 2489–2501. [Google Scholar] [CrossRef]
- Chen, Z.; Wen, W.; Bao, J.; Kuhs, K.L.; Cai, Q.; Long, J.; Shu, X.; Zheng, W.; Guo, X. Integrative Genomic Analyses of APOBEC-Mutational Signature, Expression and Germline Deletion of APOBEC3 Genes, and Immunogenicity in Multiple Cancer Types. BMC Med. Genom. 2019, 12, 131. [Google Scholar] [CrossRef]
- Wang, C.; Bai, R.; Liu, Y.; Wang, K.; Wang, Y.; Yang, J.; Cai, H.; Yang, P. Multi-Region Sequencing Depicts Intratumor Heterogeneity and Clonal Evolution in Cervical Cancer. Med. Oncol. 2023, 40, 78. [Google Scholar] [CrossRef]
- Niyazi, M.; Han, L.; Husaiyin, S.; Aishanjiang, A.; Guo, M.; Muhaimati, G.; Rozi, H.; Sun, H.; Lu, J.; Ma, C.; et al. Analysis of Somatic Mutations and Key Driving Factors of Cervical Cancer Progression. Open Med. 2023, 18, 20230759. [Google Scholar] [CrossRef]
- Periyasamy, M.; Singh, A.K.; Gemma, C.; Kranjec, C.; Farzan, R.; Leach, D.A.; Navaratnam, N.; Pálinkás, H.L.; Vértessy, B.G.; Fenton, T.R.; et al. P53 Controls Expression of the DNA Deaminase APOBEC3B to Limit Its Potential Mutagenic Activity in Cancer Cells. Nucleic Acids Res. 2017, 45, 11056–11069. [Google Scholar] [CrossRef]
- Fenton, T.R. Accumulation of Host Cell Genetic Errors Following High-Risk HPV Infection. Curr. Opin. Virol. 2021, 51, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Bournique, E.; Bowen, D.; Jalili, P.; Sanchez, A.; Ward, I.; Dananberg, A.; Manjunath, L.; Tran, G.P.; Semler, B.L.; et al. Genotoxic Stress and Viral Infection Induce Transient Expression of APOBEC3A and Pro-Inflammatory Genes through Two Distinct Pathways. Nat. Commun. 2021, 12, 4917. [Google Scholar] [CrossRef] [PubMed]
- Middlebrooks, C.D.; Banday, A.R.; Matsuda, K.; Udquim, K.-I.; Onabajo, O.O.; Paquin, A.; Figueroa, J.D.; Zhu, B.; Koutros, S.; Kubo, M.; et al. Association of Germline Variants in the APOBEC3 Region with Cancer Risk and Enrichment with APOBEC-Signature Mutations in Tumors. Nat. Genet. 2016, 48, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.J.; Reddin, I.; Policelli, P.; Oh, S.; Zainal, N.; Howes, E.; Jenkins, B.; Tracy, I.; Edmond, M.; Sharpe, B.; et al. Differentiation Signals Induce APOBEC3A Expression via GRHL3 in Squamous Epithelia and Squamous Cell Carcinoma. EMBO J. 2024, 44, 1–29. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Qin, J.; Chen, Y.; Liu, L.; Zhang, D.; Wang, M.; Wang, M.; Zhang, D. APOBEC3A Possesses Anticancer and Antiviral Effects by Differential Inhibition of HPV E6 and E7 Expression on Cervical Cancer. Int. J. Clin. Exp. Med. 2015, 8, 10548–10557. [Google Scholar]
- Cescon, D.W.; Haibe-Kains, B.; Mak, T.W. APOBEC3B Expression in Breast Cancer Reflects Cellular Proliferation, While a Deletion Polymorphism Is Associated with Immune Activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2841–2846. [Google Scholar] [CrossRef]
- Faden, D.L.; Ding, F.; Lin, Y.; Zhai, S.; Kuo, F.; Chan, T.A.; Morris, L.G.; Ferris, R.L. APOBEC Mutagenesis Is Tightly Linked to the Immune Landscape and Immunotherapy Biomarkers in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2019, 96, 140–147. [Google Scholar] [CrossRef]
- Chen, Y.J.; Roumeliotis, T.I.; Chang, Y.H.; Chen, C.T.; Han, C.L.; Lin, M.H.; Chen, H.W.; Chang, G.C.; Chang, Y.L.; Wu, C.T.; et al. Proteogenomics of Non-Smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell 2020, 182, 226–244.e17. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castilha, E.P.; Biondo, R.; Trugilo, K.P.; Fortunato, G.M.; Fenton, T.R.; Oliveira, K.B.d. APOBEC3 Proteins: From Antiviral Immunity to Oncogenic Drivers in HPV-Positive Cancers. Viruses 2025, 17, 436. https://doi.org/10.3390/v17030436
Castilha EP, Biondo R, Trugilo KP, Fortunato GM, Fenton TR, Oliveira KBd. APOBEC3 Proteins: From Antiviral Immunity to Oncogenic Drivers in HPV-Positive Cancers. Viruses. 2025; 17(3):436. https://doi.org/10.3390/v17030436
Chicago/Turabian StyleCastilha, Eliza Pizarro, Rosalba Biondo, Kleber Paiva Trugilo, Giulia Mariane Fortunato, Timothy Robert Fenton, and Karen Brajão de Oliveira. 2025. "APOBEC3 Proteins: From Antiviral Immunity to Oncogenic Drivers in HPV-Positive Cancers" Viruses 17, no. 3: 436. https://doi.org/10.3390/v17030436
APA StyleCastilha, E. P., Biondo, R., Trugilo, K. P., Fortunato, G. M., Fenton, T. R., & Oliveira, K. B. d. (2025). APOBEC3 Proteins: From Antiviral Immunity to Oncogenic Drivers in HPV-Positive Cancers. Viruses, 17(3), 436. https://doi.org/10.3390/v17030436