
Genomic Insights into Neglected Orthobunyaviruses: Molecular Characterization and Phylogenetic Analysis

 ,
,  ,
,  , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Archived Orthobunyavirus Strains Selection and Preparation
2.2. Sequencing and Bioinformatics Analyses
2.3. Phylogenetic Analysis
2.4. Prediction of Reservoir Hosts, Vector and Arthropod-Borne Vectors
3. Results
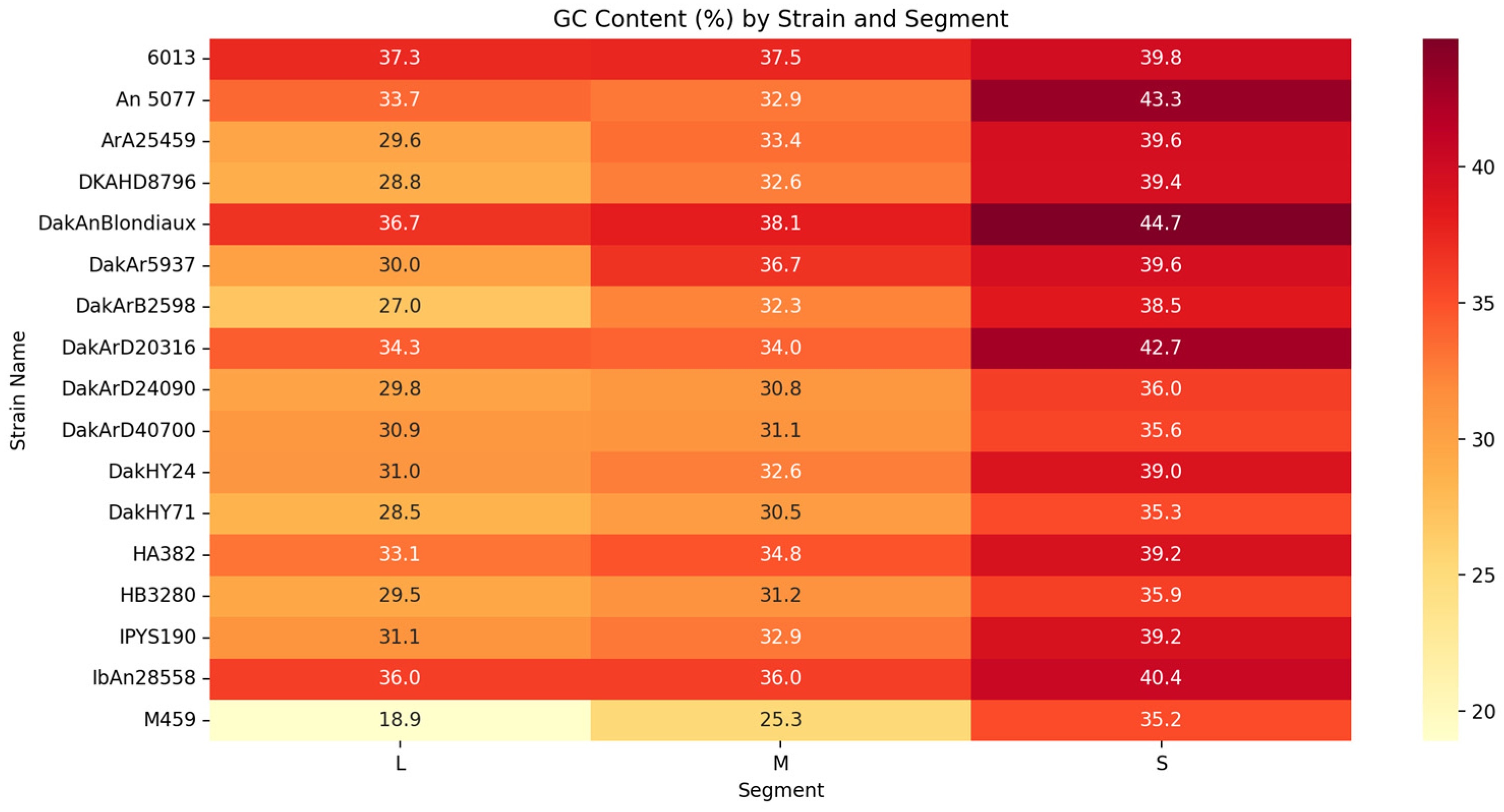
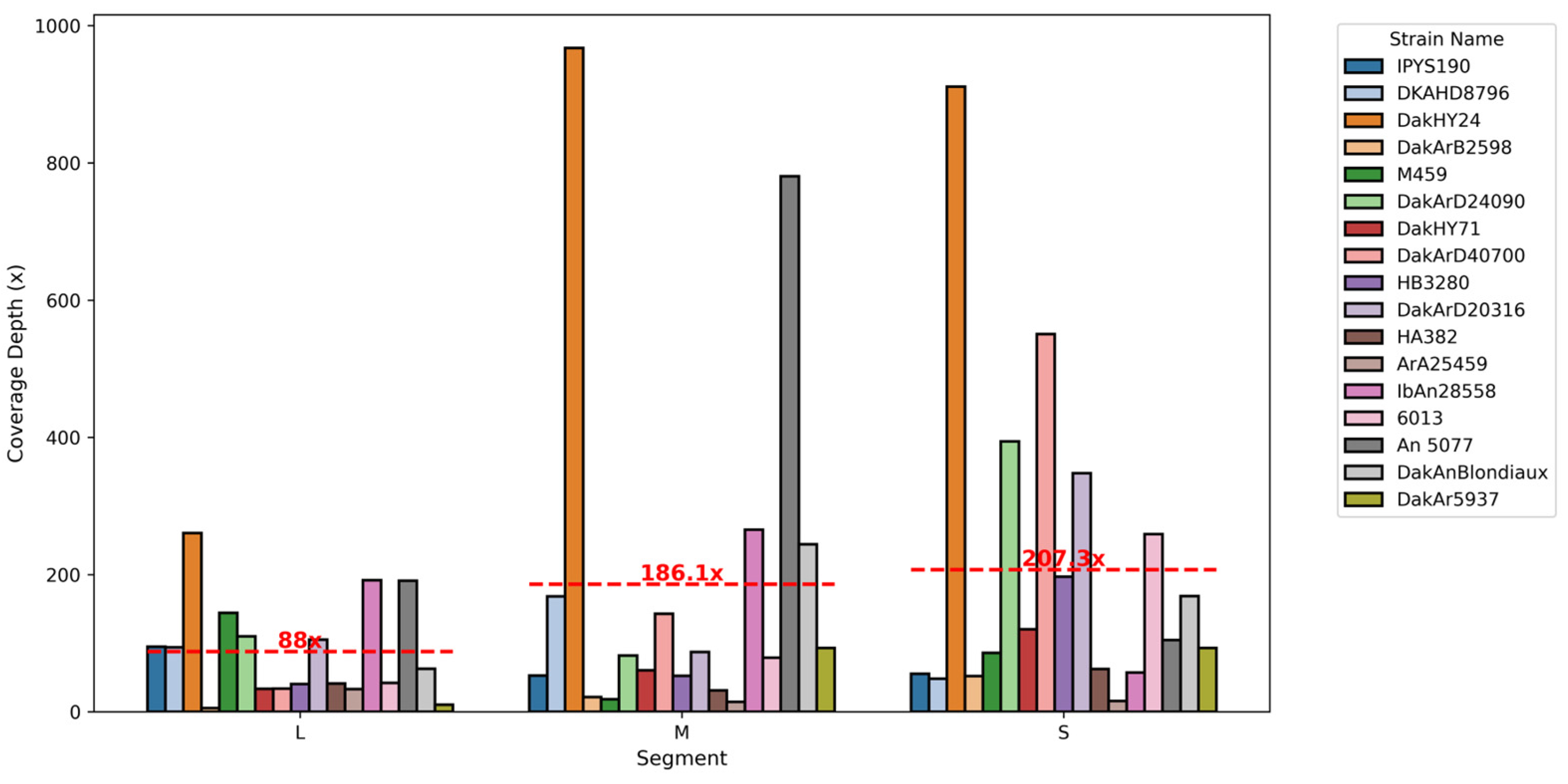
3.1. Orthobunyaviruses Sequencing Metrics
3.2. Phylogenetic Relationships and Pathogenic Connections
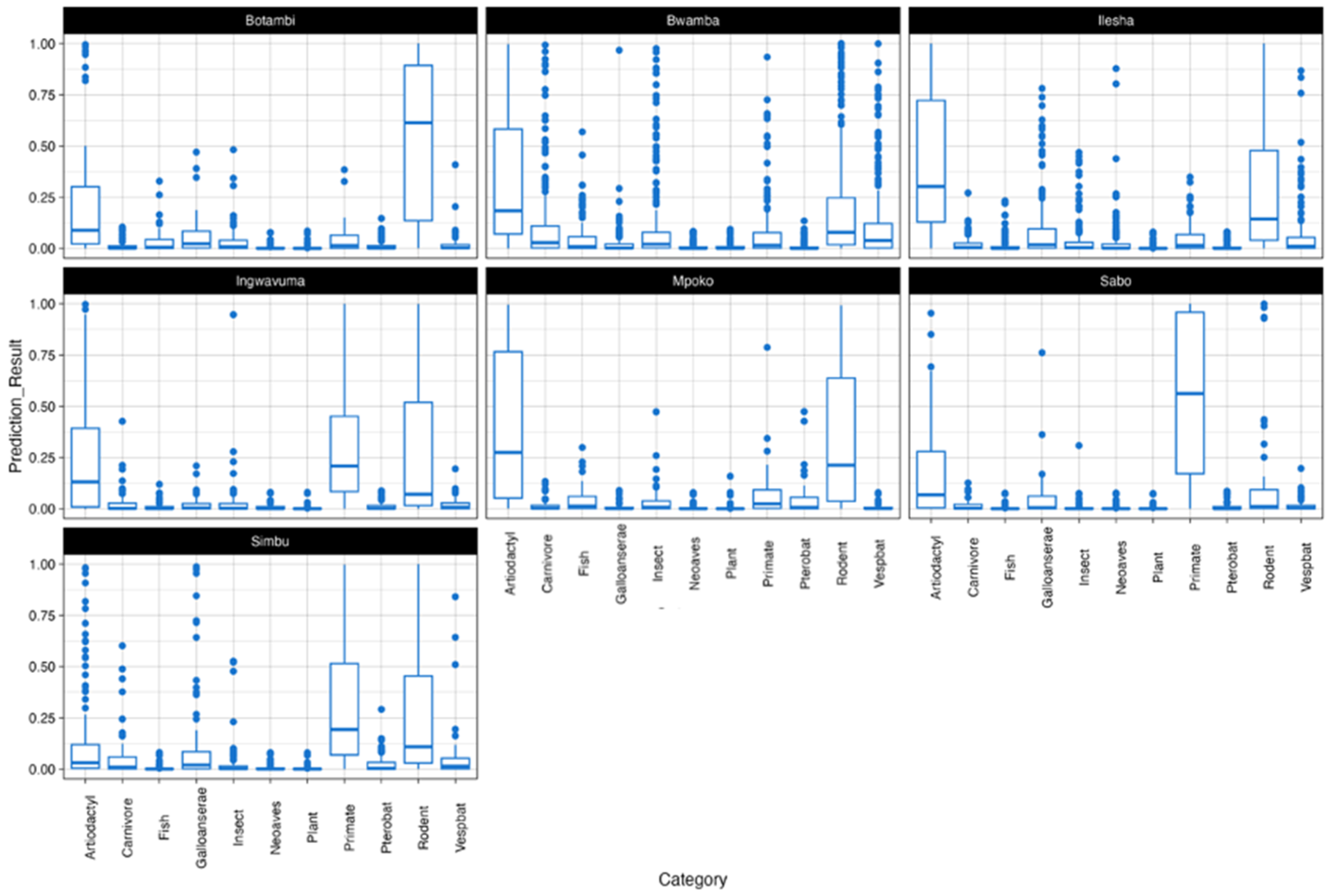
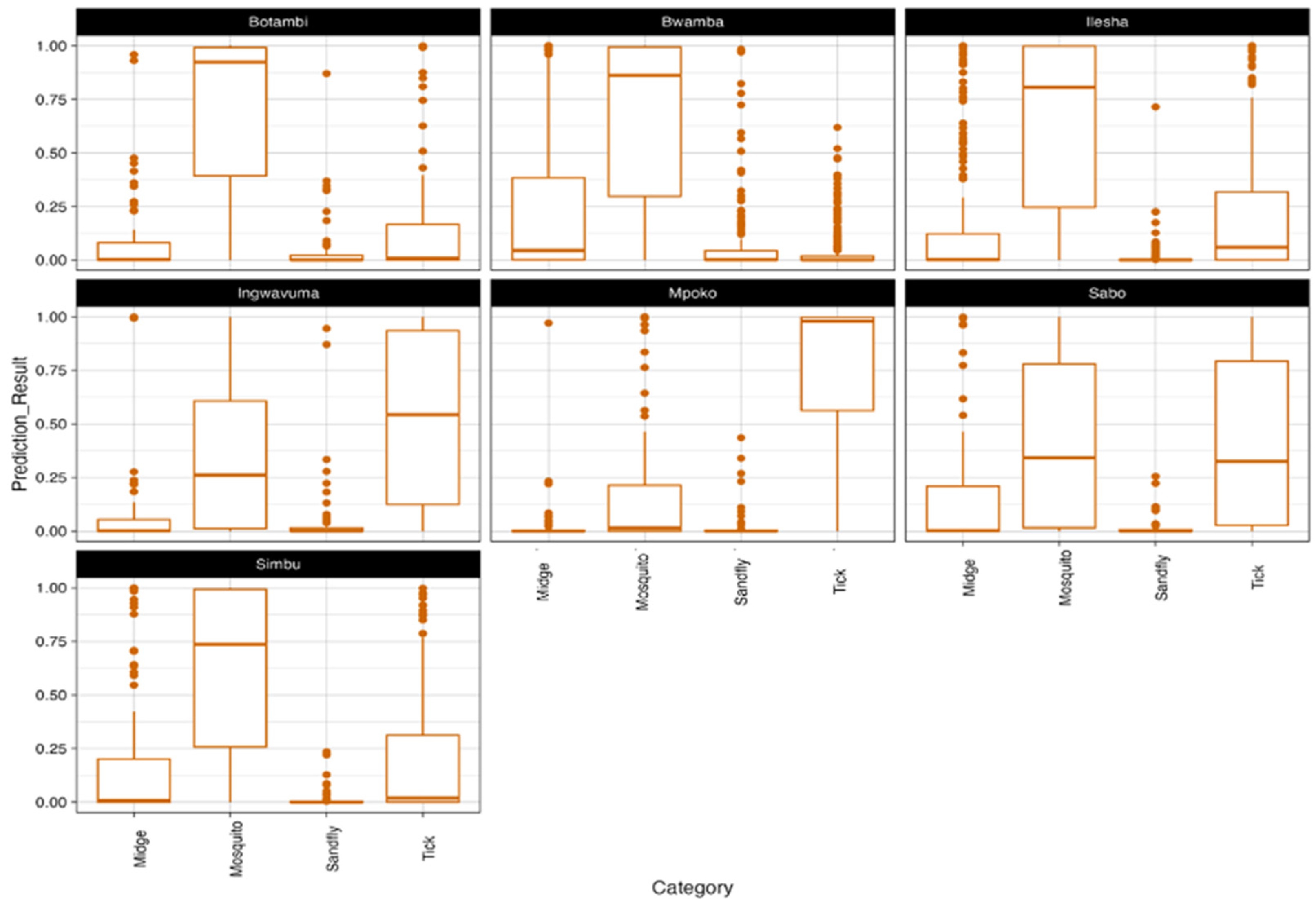
3.3. Prediction of Reservoir Hosts and Arthropod Vectors
3.3.1. Reservoir Hosts
3.3.2. Arthropod Vectors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses ICTV. [En ligne]. Disponible sur. Available online: https://ictv.global/taxonomy (accessed on 9 December 2024).
- Windhaber, S.; Xin, Q.; Lozach, P.Y. Orthobunyaviruses: From Virus Binding to Penetration into Mammalian Host Cells. Viruses 2021, 13, 872. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M. Orthobunyaviruses: Recent genetic and structural insights. Nat. Rev. Microbiol. 2014, 12, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Bird, B.; Kapoor, V.; Nichol, S.T.; Lipkin, W.I. Batai and Ngari viruses: M segment reassortment and association with severe febrile disease outbreaks in East Africa. J. Virol. 2006, 80, 5627–5630. [Google Scholar] [CrossRef] [PubMed]
- Toure, C.T.; Dieng, I.; Sankhe, S.; Kane, M.; Dia, M.; Mhamadi, M.; Ndiaye, M.; Faye, O.; Sall, A.A.; Diagne, M.M.; et al. Genomic Characterization of a Bataï Orthobunyavirus, Previously Classified as Ilesha Virus, from Field-Caught Mosquitoes in Senegal, Bandia 1969. Viruses 2024, 16, 261. [Google Scholar] [CrossRef]
- Sakkas, H.; Bozidis, P.; Franks, A.; Papadopoulou, C. Oropouche Fever: A Review. Viruses 2018, 10, 175. [Google Scholar] [CrossRef]
- Romero-Alvarez, D.; Escobar, L.E. Oropouche fever, an emergent disease from the Americas. Microbes Infect. 2018, 20, 135–146. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Wu, Z.; Feng, S.; Lu, K.; Zhu, W.; Sun, H.; Niu, G. Oropouche virus: A neglected global arboviral threat. Virus Res. 2024, 341, 199318. [Google Scholar] [CrossRef]
- Edridg, A.W.D.; van der Hoek, L. Emerging orthobunyaviruses associated with CNS disease. PLoS Negl. Trop. Dis. 2020, 14, e0008856. [Google Scholar] [CrossRef]
- Bennett, R.S.; Cress, C.M.; Ward, J.M.; Firestone, C.-Y.; Murphy, B.R.; Whitehead, S.S. La Crosse virus infectivity, pathogenesis, and immunogenicity in mice and monkeys. Virol. J. 2008, 5, 25. [Google Scholar] [CrossRef]
- Hammon, W.M.; Reeves, W.C. California encephalitis virus, a newly described agent. Calif. Med. 1952, 77, 303–309. [Google Scholar] [CrossRef]
- Gerrard, S.R.; Li, L.; Barrett, A.D.; Nichol, S.T. Ngari virus is a Bunyamwera virus reassortant that can be associated with large outbreaks of hemorrhagic fever in Africa. J. Virol. 2004, 78, 8922–8926. [Google Scholar] [CrossRef] [PubMed]
- Zeller, H.G.; Diallo, M.; Angel, G.; Traoré-Lamizana, M.; Thonnon, J.; Digoutte, J.P.; Fontenille, D. Ngari virus (Bunyaviridae: Bunyavirus). First isolation from humans in Senegal, new mosquito vectors, its epidemiology. Bull. Soc. Pathol. Exot. (1990) 1996, 89, 12–16. [Google Scholar]
- Dutuze, M.F.; Ingabire, A.; Gafarasi, I.; Uwituze, S.; Nzayirambaho, M.; Christofferson, R.C. Identification of Bunyamwera and Possible Other Orthobunyavirus Infections and Disease in Cattle during a Rift Valley Fever Outbreak in Rwanda in 2018. Am. J. Trop. Med. Hyg. 2020, 103, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.; Pachal, N.; Mascarenhas, M.; Greig, J.; Harding, S.; Young, I.; Wilhelm, B. Cache Valley virus: A scoping review of the global evidence. Zoonoses Public Health 2019, 66, 739–758. [Google Scholar] [CrossRef]
- Hughes, H.R.; Kenney, J.L.; Calvert, A.E. Cache Valley virus: An emerging arbovirus of public and veterinary health importance. J. Med. Entomol. 2023, 60, 1230–1241. [Google Scholar] [CrossRef]
- Morvan, J.M.; Digoutte, J.P.; Marsan, P.; Roux, J.F. Ilesha virus: A new aetiological agent of haemorrhagic fever in Madagascar. Trans. R. Soc. Trop. Med. Hyg. 1994, 88, 205. [Google Scholar] [CrossRef]
- Chippaux, A.; Chippaux-Hyppolite, C.; Clergeaud, P.; Brès, P. Isolation of 2 human strains of Ilesha virus in the Central African Republic. Bull. Soc. Med. Afr. Noire Lang. Fr. 1969, 14, 88–92. [Google Scholar]
- Brès, P. Recent data from serological surveys on the prevalence of arbovirus infections in Africa, with special reference to yellow fever. Bull. World Health Organ. 1970, 43, 223–267. [Google Scholar]
- Lutwama, J.J.; Rwaguma, E.B.; Nawanga, P.L.; Mukuye, A. Isolations of Bwamba virus from south central Uganda and north eastern Tanzania. Afr. Health Sci. 2002, 2, 24–28. [Google Scholar]
- Smithburn, K.C.; Mahaffy, A.F.; Paul, J.H. Bwamba Fever and Its Causative Virus. Am. J. Trop. Med. Hyg. 1941, 21, 75–90. [Google Scholar] [CrossRef]
- Top, F.H.; Kraivapan, C.; Grossman, R.A.; Rozmiarek, H.; Edelman, R.; Gould, D.J. Ingwavuma virus in Thailand. Infection of domestic pigs. Am. J. Trop. Med. Hyg. 1974, 23, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sreelekshmi, P.R.; Godke, Y.S.; Sudeep, A.B. Vector competence of three species of mosquitoes to Ingwavuma virus (Manzanilla orthobunyavirus), a new bunyavirus found circulating in India. Virusdisease 2023, 34, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Oluwayelu, D.; Wernike, K.; Adebiyi, A.; Cadmus, S.; Beer, M. Neutralizing antibodies against Simbu serogroup viruses in cattle and sheep, Nigeria, 2012–2014. BMC Vet. Res. 2018, 14, 277. [Google Scholar] [CrossRef]
- Golender, N.; Bumbarov, V.; Eldar, A.; Tov, B.E.; Kenigswald, G. Isolation of Sango viruses from Israeli symptomatic cattle. Int. J. Vet. Sci. Res. 2021, 7, 69–72. [Google Scholar]
- Causey, O.R.; Kemp, G.E.; Causey, C.E.; Lee, V.H. Isolations of Simbu-group viruses in Ibadan, Nigeria 1964–69, including the new types Sango, Shamonda, Sabo and Shuni. Ann. Trop. Med. Parasitol. 1972, 66, 357–362. [Google Scholar] [CrossRef]
- Digoutte, J.P.; Pajot, F.X.; Henderson, B.E.; Brès, P.; Luong, P.N.T. The M’Poko virus (BA 365), new prototype of arbovirus isolated in the Centrafrican Republic. Ann. Inst. Pasteur 1970, 119, 512–519. [Google Scholar]
- Okuno, T. Immunological studies relating two recently isolated viruses, Germiston virus from South Africa and Ilesha virus from West Africa, to the Bunyamwera group. Am. J. Trop. Med. Hyg. 1961, 10, 223–226. [Google Scholar] [CrossRef]
- Bob, N.S.; Barry, M.A.; Diagne, M.M.; Faye, M.; Ndione, M.H.D.; Diallo, A.; Diop, M.; Diop, B.; Faye, O.; Loucoubar, C.; et al. Detection of Rift Valley Fever Virus Lineage H From South Africa Through the Syndromic Sentinel Surveillance Network in Senegal. Open Forum Infect. Dis. 2022, 9, ofab655. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ndione, M.H.D.; Diagne, M.M.; Mencattelli, G.; Diallo, A.; Ndiaye, E.H.; Di Domenico, M.; Diallo, D.; Kane, M.; Curini, V.; Top, N.M.; et al. An amplicon-based sequencing approach for Usutu virus characterization. Virol. J. 2024, 21, 163. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Molecular Evolution, Phylogenetics and Epidemiology. Consulté le: 5 Janvier 2025. [En Ligne]. Disponible sur. Available online: http://tree.bio.ed.ac.uk/ (accessed on 9 December 2024).
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef]
- Viral Host Predictor Website. Consulté le: 9 Octobre 2024. [En ligne]. Disponible sur. Available online: http://host-predict.cvr.gla.ac.uk/ (accessed on 9 December 2024).
- R: The R Project for Statistical Computing. Consulté le: 3 Janvier 2025. [En Ligne]. Disponible sur. Available online: https://www.r-project.org/ (accessed on 9 December 2024).
- Wickham, H. Data Analysis. In ggplot2: Elegant Graphics for Data Analysis; Wickham, H., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 189–201. [Google Scholar] [CrossRef]
- Silva, J.M.; Pratas, D.; Caetano, T.; Matos, S. The complexity landscape of viral genomes. GigaScience 2022, 11, giac079. [Google Scholar] [CrossRef]
- Nagy, P.D.; Bujarski, J.J. Silencing homologous RNA recombination hot spots with GC-rich sequences in brome mosaic virus. J. Virol. 1998, 72, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, B.F.; Glaser, C.; Pedrin, R.E.; Chiles, R.E. The first reported case of California encephalitis in more than 50 years. Emerg. Infect. Dis. 2001, 7, 451–452. [Google Scholar] [CrossRef]
- Brenner, J.; Tsuda, T.; Yadin, H.; Chai, D.; Stram, Y.; Kato, T. Serological and clinical evidence of a teratogenic Simbu serogroup virus infection of cattle in Israel, 2001–2003. Vet. Ital. 2004, 40, 119–123. [Google Scholar]
- Kapuscinski, M.L.; Bergren, N.A.; Russell, B.J.; Lee, J.S.; Borland, E.M.; Hartman, D.A.; King, D.C.; Hughes, H.R.; Burkhalter, K.L.; Kading, R.C.; et al. Genomic characterization of 99 viruses from the bunyavirus families Nairoviridae, Peribunyaviridae, and Phenuiviridae, including 35 previously unsequenced viruses. PLoS Pathog. 2021, 17, e1009315. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name | Viral Species | First Identification (Place and Year) | Reported Human Case | Geographical Distribution | Vectors/Isolation Species | Clinical Manifestations |
|---|---|---|---|---|---|---|
| Bwamba Virus (BWAV) | Orthobunyavirus bwambaense | Uganda in 1937 [20] | Yes | Uganda, Central African Republic, South Africa, Cameroun, Senegal | Anopheles gambiae Anopheles funestus | Exanthem with meningeal involvement, fever, headache, arthralgia, diarrhea, body rash. |
| Ilesha Virus (ILEV) | Orthobunyavirus ileshaense | Nigeria in 1957 [28] | Yes | Central African Republic, Cameroun, Madagascar, Senegal | Anopheles gambiae | Fatal meningoencephalitis, hemorrhagic fever |
| Ingwavuma Virus (INGV) | Orthobunyavirus ingwavumaense | Thailande in 1970 [22] | Yes | Indonesia, India, Thailand, Nigeria | Culex vishnui Hyphanturgus ocularius | Febrile illnesses |
| Simbu Virus (SIMV) | Orthobunyavirus simbuense | Nigeria in 1964–1969 [26] | No | South Africa, Nigeria, Israel | Mosquito and midge | Unknown |
| Sango Virus (SANV) | Orthobunyavirus sangoense | Nigeria in 1965 [26] | No | Nigeria, Israel | Culocoides spp. Blood cattle | Unknown |
| Sabo Virus (SABOV) | Orthobunyavirus saboense | Nigeria in 1966 [26] | No | Nigeria, Central African Republic | Blood goat | Unknown |
| M’Poko Virus (MPOV) | Orthobunyavirus mpokoense | Central African Republic in 1966 [27] | No | Central African Republic, Senegal | Culex pruina Culex weschei Culex perfuscus) | Unknown |
| Botambi Virus (BOTV) | Orthobunyavirus botambiense | Central African Republic in 1968 [27] | No | Central African Republic, Cote d’Ivoire | Culex guiarti | Unknown |
| Tanga Virus (TANV) | Orthobunyavirus tangaense | Tanzania in 1962 [27] | No | Tanzania, Burkina Faso, Cote d’Ivoire | Anopheles funestus | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sankhe, S.; Dieng, I.; Kane, M.; Diallo, A.; Ndiaye, N.A.; Top, N.M.; Dia, M.; Faye, O.; Sall, A.A.; Faye, O.; et al. Genomic Insights into Neglected Orthobunyaviruses: Molecular Characterization and Phylogenetic Analysis. Viruses 2025, 17, 406. https://doi.org/10.3390/v17030406
Sankhe S, Dieng I, Kane M, Diallo A, Ndiaye NA, Top NM, Dia M, Faye O, Sall AA, Faye O, et al. Genomic Insights into Neglected Orthobunyaviruses: Molecular Characterization and Phylogenetic Analysis. Viruses. 2025; 17(3):406. https://doi.org/10.3390/v17030406
Chicago/Turabian StyleSankhe, Safiétou, Idrissa Dieng, Mouhamed Kane, Amadou Diallo, Ndeye Awa Ndiaye, Ndeye Marieme Top, Moussa Dia, Ousmane Faye, Amadou Alpha Sall, Oumar Faye, and et al. 2025. "Genomic Insights into Neglected Orthobunyaviruses: Molecular Characterization and Phylogenetic Analysis" Viruses 17, no. 3: 406. https://doi.org/10.3390/v17030406
APA StyleSankhe, S., Dieng, I., Kane, M., Diallo, A., Ndiaye, N. A., Top, N. M., Dia, M., Faye, O., Sall, A. A., Faye, O., Sembene, P. M., Loucoubar, C., Faye, M., & Diagne, M. M. (2025). Genomic Insights into Neglected Orthobunyaviruses: Molecular Characterization and Phylogenetic Analysis. Viruses, 17(3), 406. https://doi.org/10.3390/v17030406