
Coevolution of Lentiviral Vif with Host A3F and A3G: Insights from Computational Modelling and Ancestral Sequence Reconstruction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
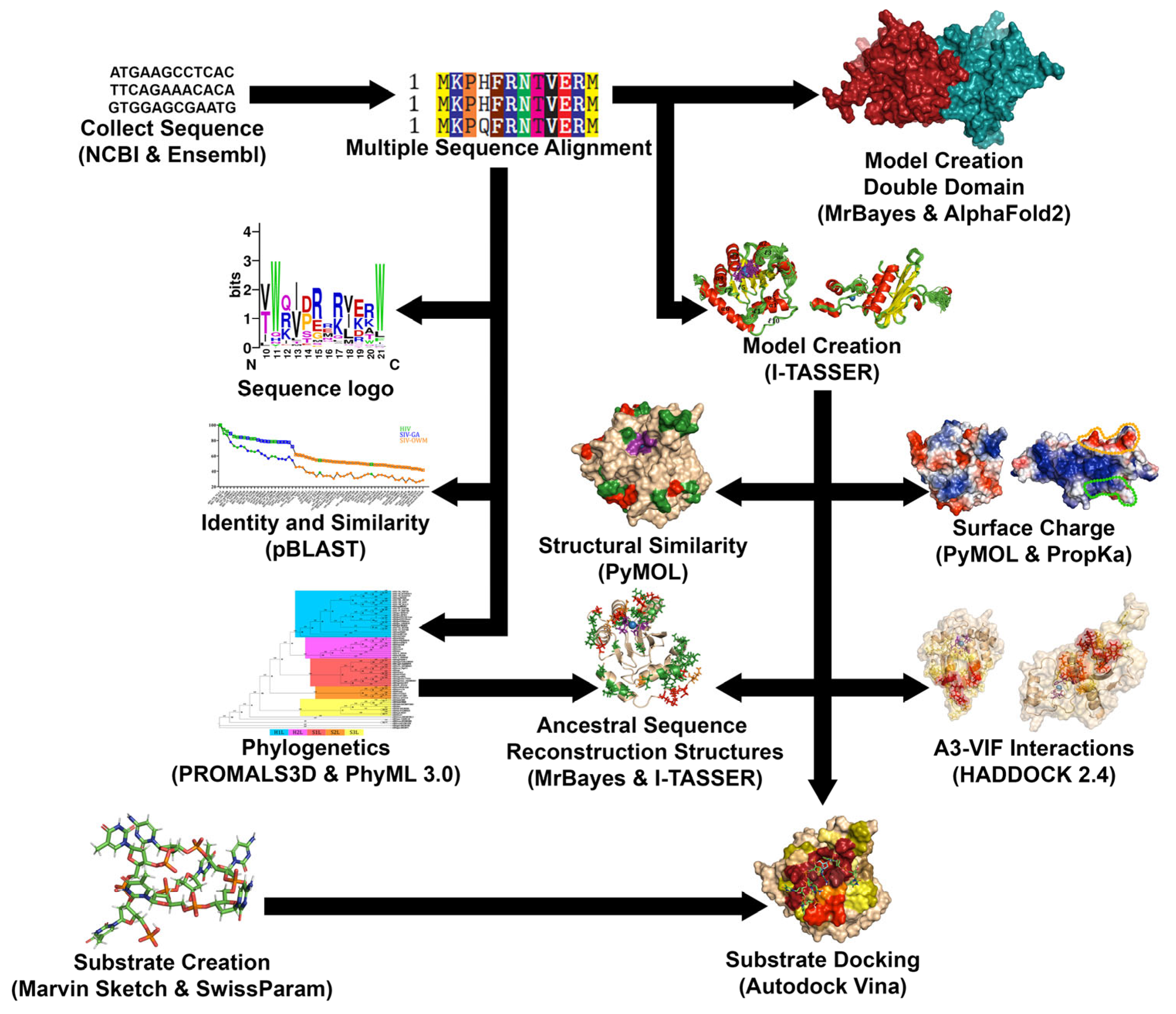
2. Materials and Methods
2.1. Acquisition and Alignment of Sequences
2.2. Production of Similarity Graphs and Phylogenetic Trees
2.3. Creation of 3D Structural Homology Models
2.4. Analysis of Structural Surfaces and Charge
2.5. Protein-Protein Docking
2.6. Creation of DNA Substrates
2.7. Analysis of DNA-Protein Docking Results
2.8. Ancestral Sequence Reconstruction
3. Results
3.1. Evolution of A3F, A3G, and Vif
3.1.1. Vif Contains More Variation than A3FGs in Its Binding Regions
3.1.2. Rapid Genetic Evolution of Vif and A3FGs
3.1.3. Overall Structure of Vif from HIV Lineages and Primate A3FGs Maintained
3.2. Structural Evolution of A3F and A3G
3.2.1. The Amino Acid Variation of APOBEC3F and APOBEC3G
3.2.2. Structural Variation of A3F and A3G Across the Species
3.2.3. The Role of Charge Variations in Vif and A3 Proteins
3.3. Overall A3 and Vif-Binding Modes Remain Similar Despite Adaptation
3.4. A3FG-DNA Interactions
3.4.1. DNA Conformations and the Catalytic Pocket of A3F and A3G
3.4.2. Molecular Interactions of DNA Sequences with APOBEC3F
3.4.3. Molecular Interactions of DNA Sequences with APOBEC3G
3.4.4. Comparison of A3F and A3G Double Domains Across Ancestral Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APOBEC | Apolipoprotein B mRNA editing polypeptide like |
| A3F | APOBEC3F |
| A3G | APOBEC3G |
| Vif | Viral infectivity factor |
| GAs | Great apes |
| OWMs | Old World monkeys |
| NWMs | New World monkeys |
References
- Kluge, S.F.; Sauter, D.; Kirchhoff, F. SnapShot: Antiviral restriction factors. Cell 2015, 163, 774–774 e1. [Google Scholar] [CrossRef] [PubMed]
- Kirmaier, A.; Krupp, A.; Johnson, W.E. Understanding restriction factors and intrinsic immunity: Insights and lessons from the primate lentiviruses. Future Virol. 2014, 9, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Desimmie, B.A.; Pathak, V.K. Structural Insights into APOBEC3-Mediated Lentiviral Restriction. Viruses 2020, 12, 587. [Google Scholar] [CrossRef]
- Nakano, Y.; Aso, H.; Soper, A.; Yamada, E.; Moriwaki, M.; Juarez-Fernandez, G.; Koyanagi, Y.; Sato, K. A conflict of interest: The evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology 2017, 14, 31. [Google Scholar] [CrossRef]
- Sauter, D.; Kirchhoff, F. Key Viral Adaptations Preceding the AIDS Pandemic. Cell Host Microbe 2019, 25, 27–38. [Google Scholar] [CrossRef]
- Song, B.; Javanbakht, H.; Perron, M.; Park, D.H.; Stremlau, M.; Sodroski, J. Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J. Virol. 2005, 79, 3930–3937. [Google Scholar] [CrossRef]
- Marx, P.A.; Apetrei, C.; Drucker, E. AIDS as a zoonosis? Confusion over the origin of the virus and the origin of the epidemics. J. Med. Primatol. 2004, 33, 220–226. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef]
- Ahuka-Mundeke, S.; Ayouba, A.; Mbala-Kingebeni, P.; Liegeois, F.; Esteban, A.; Lunguya-Metila, O.; Demba, D.; Bilulu, G.; Mbenzo-Abokome, V.; Inogwabini, B.I.; et al. Novel multiplexed HIV/simian immunodeficiency virus antibody detection assay. Emerg. Infect. Dis. 2011, 17, 2277–2286. [Google Scholar] [CrossRef]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pepin, J.; et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Silvestri, G.; Hirsch, V. Nonpathogenic simian immunodeficiency virus infections. Cold Spring Harb. Perspect. Med. 2012, 2, a007153. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, S.U.; Chen, K.; Bhagwat, A.S. Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases. Chem. Rev. 2016, 116, 12688–12710. [Google Scholar] [CrossRef] [PubMed]
- Meulendyke, K.A.; Croteau, J.D.; Zink, M.C. HIV life cycle, innate immunity and autophagy in the central nervous system. Curr. Opin. HIV AIDS 2014, 9, 565–571. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef]
- Song, C.; Sutton, L.; Johnson, M.E.; D’Aquila, R.T.; Donahue, J.P. Signals in APOBEC3F N-terminal and C-terminal deaminase domains each contribute to encapsidation in HIV-1 virions and are both required for HIV-1 restriction. J. Biol. Chem. 2012, 287, 16965–16974. [Google Scholar] [CrossRef]
- Feng, Y.; Baig, T.T.; Love, R.P.; Chelico, L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 2014, 5, 450. [Google Scholar] [CrossRef]
- Feng, Y.; Love, R.P.; Ara, A.; Baig, T.T.; Adolph, M.B.; Chelico, L. Natural Polymorphisms and Oligomerization of Human APOBEC3H Contribute to Single-stranded DNA Scanning Ability. J. Biol. Chem. 2015, 290, 27188–27203. [Google Scholar] [CrossRef]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef]
- Mack, K.; Starz, K.; Sauter, D.; Langer, S.; Bibollet-Ruche, F.; Learn, G.H.; Sturzel, C.M.; Leoz, M.; Plantier, J.C.; Geyer, M.; et al. Efficient Vpu-Mediated Tetherin Antagonism by an HIV-1 Group O Strain. J. Virol. 2017, 91, 20. [Google Scholar] [CrossRef] [PubMed]
- Firrito, C.; Bertelli, C.; Vanzo, T.; Chande, A.; Pizzato, M. SERINC5 as a New Restriction Factor for Human Immunodeficiency Virus and Murine Leukemia Virus. Annu. Rev. Virol. 2018, 5, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, D.; Rai, M.; Gaur, R. Novel host restriction factors implicated in HIV-1 replication. J. Gen. Virol. 2018, 99, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Jeang, K.T.; Tokunaga, K. Host restriction factors in retroviral infection: Promises in virus-host interaction. Retrovirology 2012, 9, 112. [Google Scholar] [CrossRef]
- Jia, X.; Zhao, Q.; Xiong, Y. HIV suppression by host restriction factors and viral immune evasion. Curr. Opin. Struct. Biol. 2015, 31, 106–114. [Google Scholar] [CrossRef]
- Wu, F.; Ourmanov, I.; Riddick, N.; Matsuda, K.; Whitted, S.; Plishka, R.J.; Buckler-White, A.; Starost, M.F.; Hirsch, V.M. TRIM5alpha restriction affects clinical outcome and disease progression in simian immunodeficiency virus-infected rhesus macaques. J. Virol. 2015, 89, 2233–2240. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, L.; Yu, Y.; Wu, J.; Yang, F.; Xu, Y.; Guo, Q.; Wu, X.; Cao, S.; Cao, L.; et al. Involvement of SAMHD1 in dNTP homeostasis and the maintenance of genomic integrity and oncotherapy (Review). Int. J. Oncol. 2020, 56, 879–888. [Google Scholar] [CrossRef]
- Coggins, S.A.; Mahboubi, B.; Schinazi, R.F.; Kim, B. SAMHD1 Functions and Human Diseases. Viruses 2020, 12, 382. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Wu, L. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol. 2019, 27, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Borzooee, F.; Asgharpour, M.; Quinlan, E.; Grant, M.D.; Larijani, M. Viral subversion of APOBEC3s: Lessons for anti-tumor immunity and tumor immunotherapy. Int. Rev. Immunol. 2018, 37, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Harris, R.S.; Harki, D.A. APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem. Biol. 2018, 25, 36–49. [Google Scholar] [CrossRef]
- Morse, M.; Huo, R.; Feng, Y.; Rouzina, I.; Chelico, L.; Williams, M.C. Dimerization regulates both deaminase-dependent and deaminase-independent HIV-1 restriction by APOBEC3G. Nat. Commun. 2017, 8, 597. [Google Scholar] [CrossRef]
- Chelico, L.; Pham, P.; Calabrese, P.; Goodman, M.F. APOBEC3G DNA deaminase acts processively 3′ --> 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006, 13, 392–399. [Google Scholar] [CrossRef]
- Monajemi, M.; Woodworth, C.F.; Zipperlen, K.; Gallant, M.; Grant, M.D.; Larijani, M. Positioning of APOBEC3G/F mutational hotspots in the human immunodeficiency virus genome favors reduced recognition by CD8+ T cells. PLoS ONE 2014, 9, e93428. [Google Scholar] [CrossRef]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Renner, T.M.; Belanger, K.; Goodwin, L.R.; Campbell, M.; Langlois, M.A. Characterization of molecular attributes that influence LINE-1 restriction by all seven human APOBEC3 proteins. Virology 2018, 520, 127–136. [Google Scholar] [CrossRef]
- OhAinle, M.; Kerns, J.A.; Li, M.M.; Malik, H.S.; Emerman, M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008, 4, 249–259. [Google Scholar] [CrossRef]
- Wang, J.; Shaban, N.M.; Land, A.M.; Brown, W.L.; Harris, R.S. Simian Immunodeficiency Virus Vif and Human APOBEC3B Interactions Resemble Those between HIV-1 Vif and Human APOBEC3G. J. Virol. 2018, 92, 14. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.; Meier, S.; MacCarthy, T. The cytidine deaminase under-representation reporter (CDUR) as a tool to study evolution of sequences under deaminase mutational pressure. BMC Bioinform. 2018, 19, 163. [Google Scholar] [CrossRef]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef]
- Holland, S.J.; Berghuis, L.M.; King, J.J.; Iyer, L.M.; Sikora, K.; Fifield, H.; Peter, S.; Quinlan, E.M.; Sugahara, F.; Shingate, P.; et al. Expansions, diversification, and interindividual copy number variations of AID/APOBEC family cytidine deaminase genes in lampreys. Proc. Natl. Acad. Sci. USA 2018, 115, E3211–E3220. [Google Scholar] [CrossRef]
- Conticello, S.G.; Langlois, M.A.; Yang, Z.; Neuberger, M.S. DNA deamination in immunity: AID in the context of its APOBEC relatives. Adv. Immunol. 2007, 94, 37–73. [Google Scholar] [CrossRef]
- Barreto, V.M.; Magor, B.G. Activation-induced cytidine deaminase structure and functions: A species comparative view. Dev. Comp. Immunol. 2011, 35, 991–1007. [Google Scholar] [CrossRef]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2, E275. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Davidson, N.O.; Cho, S.J.; Malim, M.H. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004, 14, 1392–1396. [Google Scholar] [CrossRef]
- Compton, A.A.; Emerman, M. Convergence and divergence in the evolution of the APOBEC3G-Vif interaction reveal ancient origins of simian immunodeficiency viruses. PLoS Pathog. 2013, 9, e1003135. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Hirsch, V.M.; Emerman, M. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe 2012, 11, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Krupp, A.; McCarthy, K.R.; Ooms, M.; Letko, M.; Morgan, J.S.; Simon, V.; Johnson, W.E. APOBEC3G polymorphism as a selective barrier to cross-species transmission and emergence of pathogenic SIV and AIDS in a primate host. PLoS Pathog. 2013, 9, e1003641. [Google Scholar] [CrossRef] [PubMed]
- Adolph, M.B.; Love, R.P.; Feng, Y.; Chelico, L. Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 2017, 45, 11925–11940. [Google Scholar] [CrossRef]
- Chen, H.; Lilley, C.E.; Yu, Q.; Lee, D.V.; Chou, J.; Narvaiza, I.; Landau, N.R.; Weitzman, M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006, 16, 480–485. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef]
- Ito, S.; Nagaoka, H.; Shinkura, R.; Begum, N.; Muramatsu, M.; Nakata, M.; Honjo, T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Natl. Acad. Sci. USA 2004, 101, 1975–1980. [Google Scholar] [CrossRef]
- Wichroski, M.J.; Robb, G.B.; Rana, T.M. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2006, 2, e41. [Google Scholar] [CrossRef]
- Bouzidi, M.S.; Caval, V.; Suspene, R.; Hallez, C.; Pineau, P.; Wain-Hobson, S.; Vartanian, J.P. APOBEC3DE Antagonizes Hepatitis B Virus Restriction Factors APOBEC3F and APOBEC3G. J. Mol. Biol. 2016, 428, 3514–3528. [Google Scholar] [CrossRef]
- Liang, W.; Xu, J.; Yuan, W.; Song, X.; Zhang, J.; Wei, W.; Yu, X.F.; Yang, Y. APOBEC3DE Inhibits LINE-1 Retrotransposition by Interacting with ORF1p and Influencing LINE Reverse Transcriptase Activity. PLoS ONE 2016, 11, e0157220. [Google Scholar] [CrossRef]
- Feng, Y.; Goubran, M.H.; Follack, T.B.; Chelico, L. Deamination-independent restriction of LINE-1 retrotransposition by APOBEC3H. Sci. Rep. 2017, 7, 10881. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Adolph, M.B.; Love, R.P.; Chelico, L. Biochemical Basis of APOBEC3 Deoxycytidine Deaminase Activity on Diverse DNA Substrates. ACS Infect. Dis. 2018, 4, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Emerman, M.; Malik, H.S.; McLaughlin, R.N.J. Retrocopying expands the functional repertoire of APOBEC3 antiviral proteins in primates. eLife 2020, 9, e58436. [Google Scholar] [CrossRef]
- Yang, H.; Kim, K.; Li, S.; Pacheco, J.; Chen, X.S. Structural basis of sequence-specific RNA recognition by the antiviral factor APOBEC3G. Nat. Commun. 2022, 13, 7498. [Google Scholar] [CrossRef]
- Ito, F.; Alvarez-Cabrera, A.L.; Liu, S.; Yang, H.; Shiriaeva, A.; Zhou, Z.H.; Chen, X.S. Structural basis for HIV-1 antagonism of host APOBEC3G via Cullin E3 ligase. Sci. Adv. 2023, 9, eade3168. [Google Scholar] [CrossRef]
- Kouno, T.; Shibata, S.; Shigematsu, M.; Hyun, J.; Kim, T.G.; Matsuo, H.; Wolf, M. Structural insights into RNA bridging between HIV-1 Vif and antiviral factor APOBEC3G. Nat. Commun. 2023, 14, 4037. [Google Scholar] [CrossRef]
- Adolph, M.B.; Webb, J.; Chelico, L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. PLoS ONE 2013, 8, e64196. [Google Scholar] [CrossRef]
- Ziegler, S.J.; Liu, C.; Landau, M.; Buzovetsky, O.; Desimmie, B.A.; Zhao, Q.; Sasaki, T.; Burdick, R.C.; Pathak, V.K.; Anderson, K.S.; et al. Insights into DNA substrate selection by APOBEC3G from structural, biochemical, and functional studies. PLoS ONE 2018, 13, e0195048. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Gunthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2017, 24, 132–133. [Google Scholar] [CrossRef]
- Nisole, S.; Saib, A. Early steps of retrovirus replicative cycle. Retrovirology 2004, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Love, R.P.; Chelico, L. HIV-1 viral infectivity factor (Vif) alters processive single-stranded DNA scanning of the retroviral restriction factor APOBEC3G. J. Biol. Chem. 2013, 288, 6083–6094. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Chen, D.; Landau, N.R. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [CrossRef]
- Ara, A.; Love, R.P.; Chelico, L. Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PLoS Pathog. 2014, 10, e1004024. [Google Scholar] [CrossRef]
- Ara, A.; Love, R.P.; Follack, T.B.; Ahmed, K.A.; Adolph, M.B.; Chelico, L. Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G. J. Virol. 2017, 91, 19. [Google Scholar] [CrossRef]
- Feng, Y.; Chelico, L. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J. Biol. Chem. 2011, 286, 11415–11426. [Google Scholar] [CrossRef]
- Bohn, M.F.; Shandilya, S.M.; Albin, J.S.; Kouno, T.; Anderson, B.D.; McDougle, R.M.; Carpenter, M.A.; Rathore, A.; Evans, L.; Davis, A.N.; et al. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding domain. Structure 2013, 21, 1042–1050. [Google Scholar] [CrossRef]
- Siu, K.K.; Sultana, A.; Azimi, F.C.; Lee, J.E. Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 2013, 4, 2593. [Google Scholar] [CrossRef]
- Nakashima, M.; Ode, H.; Kawamura, T.; Kitamura, S.; Naganawa, Y.; Awazu, H.; Tsuzuki, S.; Matsuoka, K.; Nemoto, M.; Hachiya, A.; et al. Structural Insights into HIV-1 Vif-APOBEC3F Interaction. J. Virol. 2016, 90, 1034–1047. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xiao, X.; Li, S.X.; Wolfe, A.; Chen, X.S. Molecular Interactions of a DNA Modifying Enzyme APOBEC3F Catalytic Domain with a Single-Stranded DNA. J. Mol. Biol. 2018, 430, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Kouno, T.; Luengas, E.M.; Shigematsu, M.; Shandilya, S.M.; Zhang, J.; Chen, L.; Hara, M.; Schiffer, C.A.; Harris, R.S.; Matsuo, H. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 2015, 22, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, S.X.; Yang, H.; Chen, X.S. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 2016, 7, 12193. [Google Scholar] [CrossRef]
- Holden, L.G.; Prochnow, C.; Chang, Y.P.; Bransteitter, R.; Chelico, L.; Sen, U.; Stevens, R.C.; Goodman, M.F.; Chen, X.S. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 2008, 456, 121–124. [Google Scholar] [CrossRef]
- Shandilya, S.M.; Nalam, M.N.; Nalivaika, E.A.; Gross, P.J.; Valesano, J.C.; Shindo, K.; Li, M.; Munson, M.; Royer, W.E.; Harjes, E.; et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 2010, 18, 28–38. [Google Scholar] [CrossRef]
- Shandilya, S.M.; Bohn, M.F.; Schiffer, C.A. A computational analysis of the structural determinants of APOBEC3’s catalytic activity and vulnerability to HIV-1 Vif. Virology 2014, 471–473, 105–116. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, T.; Xu, Z.; Liu, S.; Zhao, B.; Lan, W.; Wang, C.; Ding, J.; Cao, C. Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J. Biol. Chem. 2015, 290, 4010–4021. [Google Scholar] [CrossRef]
- Maiti, A.; Myint, W.; Kanai, T.; Delviks-Frankenberry, K.; Sierra Rodriguez, C.; Pathak, V.K.; Schiffer, C.A.; Matsuo, H. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018, 9, 2460. [Google Scholar] [CrossRef]
- Wan, L.; Nagata, T.; Katahira, M. Influence of the DNA sequence/length and pH on deaminase activity, as well as the roles of the amino acid residues around the catalytic center of APOBEC3F. Phys. Chem. Chem. Phys. 2018, 20, 3109–3117. [Google Scholar] [CrossRef]
- Gorle, S.; Pan, Y.; Sun, Z.; Shlyakhtenko, L.S.; Harris, R.S.; Lyubchenko, Y.L.; Vukovic, L. Computational Model and Dynamics of Monomeric Full-Length APOBEC3G. ACS Cent. Sci. 2017, 3, 1180–1188. [Google Scholar] [CrossRef]
- Ewald, J.; Sieber, P.; Garde, R.; Lang, S.N.; Schuster, S.; Ibrahim, B. Trends in mathematical modeling of host-pathogen interactions. Cell. Mol. Life Sci. 2020, 77, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Wang, L.; Meng, F.L.; Hwang, J.K.; Alt, F.W.; Wu, H. AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell 2017, 67, 361–373 e364. [Google Scholar] [CrossRef] [PubMed]
- King, J.J.; Larijani, M. A Novel Regulator of Activation-Induced Cytidine Deaminase/APOBECs in Immunity and Cancer: Schrodinger’s CATalytic Pocket. Front. Immunol. 2017, 8, 351. [Google Scholar] [CrossRef] [PubMed]
- King, J.J.; Manuel, C.A.; Barrett, C.V.; Raber, S.; Lucas, H.; Sutter, P.; Larijani, M. Catalytic pocket inaccessibility of activation-induced cytidine deaminase is a safeguard against excessive mutagenic activity. Structure 2015, 23, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef]
- Gille, C.; Fahling, M.; Weyand, B.; Wieland, T.; Gille, A. Alignment-Annotator web server: Rendering and annotating sequence alignments. Nucleic Acids Res. 2014, 42, W3–W6. [Google Scholar] [CrossRef]
- Gille, C.; Frommel, C. STRAP: Editor for STRuctural Alignments of Proteins. Bioinformatics 2001, 17, 377–378. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Madden, T. The BLAST Sequence Analysis Tool. In The NCBI Handbook [Internet], 2nd ed.; NCBI: Bethesda, MD, USA, 2013. [Google Scholar]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Shaban, N.M.; Shi, K.; Li, M.; Aihara, H.; Harris, R.S. 1.92 Angstrom Zinc-Free APOBEC3F Catalytic Domain Crystal Structure. J. Mol. Biol. 2016, 428, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef]
- Binning, J.M.; Chesarino, N.M.; Emerman, M.; Gross, J.D. Structural Basis for a Species-Specific Determinant of an SIV Vif Protein toward Hominid APOBEC3G Antagonism. Cell Host Microbe 2019, 26, 739–747 e734. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Nickle, D.C.; Heath, L.; Jensen, M.A.; Gilbert, P.B.; Mullins, J.I.; Kosakovsky Pond, S.L. HIV-specific probabilistic models of protein evolution. PLoS ONE 2007, 2, e503. [Google Scholar] [CrossRef]
- Simon, T. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. S1), S162–S173. [Google Scholar] [CrossRef]
- Benkert, P.; Tosatto, S.C.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, W.; He, B.; Walker, S.E.; Zhang, H.; Govindarajoo, B.; Virtanen, J.; Xue, Z.; Shen, H.B.; Zhang, Y. Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade. Proteins 2016, 84 (Suppl. S1), 233–246. [Google Scholar] [CrossRef]
- Kitamura, S.; Ode, H.; Nakashima, M.; Imahashi, M.; Naganawa, Y.; Kurosawa, T.; Yokomaku, Y.; Yamane, T.; Watanabe, N.; Suzuki, A.; et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012, 19, 1005–1010. [Google Scholar] [CrossRef]
- Kitamura, S.; Ode, H.; Nakashima, M.; Imahashi, M.; Naganawa, Y.; Ibe, S.; Yokomaku, Y.; Watanabe, N.; Suzuki, A.; Sugiura, W.; et al. Crystal structure of the human APOBEC3C having HIV-1 Vif-binding interface. Protein Data Bank 2011. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, T.; Wang, C.; Lan, W.; Ding, J.; Cao, C. Crystal Structure of Cytidine Deaminase Human APOBEC3F Chimeric Catalytic Domain in Complex with DNA. Chin. J. Chem. 2019, 36, 1241–1248. [Google Scholar] [CrossRef]
- Li, M.; Shandilya, S.M.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar] [CrossRef]
- Hu, Y.; Desimmie, B.A.; Nguyen, H.C.; Ziegler, S.J.; Cheng, T.C.; Chen, J.; Wang, J.; Wang, H.; Zhang, K.; Pathak, V.K.; et al. Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat. Struct. Mol. Biol. 2019, 26, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.7.6 Schrödinger, LLC. Available online: https://www.pymol.org/ (accessed on 30 September 2016).
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi,psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Diamond, C.P.; Im, J.; Button, E.A.; Huebert, D.N.G.; King, J.J.; Borzooee, F.; Abdouni, H.S.; Bacque, L.; McCarthy, E.; Fifield, H.; et al. AID, APOBEC3A and APOBEC3B efficiently deaminate deoxycytidines neighboring DNA damage induced by oxidation or alkylation. Biochim. Et Biophys. Acta. Gen. Subj. 2019, 1863, 129415. [Google Scholar] [CrossRef]
- Ghorbani, A.; Khataeipour, S.J.; Solbakken, M.H.; Huebert, D.N.G.; Khoddami, M.; Eslamloo, K.; Collins, C.; Hori, T.; Jentoft, S.; Rise, M.L.; et al. Ancestral reconstruction reveals catalytic inactivation of activation-induced cytidine deaminase concomitant with cold water adaption in the Gadiformes bony fish. BMC Biol. 2022, 20, 293. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.J.; Ahn, J.; Mitra, M.; Byeon, C.H.; Hercik, K.; Hritz, J.; Charlton, L.M.; Levin, J.G.; Gronenborn, A.M. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013, 4, 1890. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.J.; Byeon, C.H.; Wu, T.; Mitra, M.; Singer, D.; Levin, J.G.; Gronenborn, A.M. Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 2016, 55, 2944–2959. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.; Afif, S.A.; Shimoda, M.; Maeda, K.; Sakaguchi, N.; Pedersen, L.C.; Goodman, M.F. Structural analysis of the activation-induced deoxycytidine deaminase required in immunoglobulin diversification. DNA Repair 2016, 43, 48–56. [Google Scholar] [CrossRef]
- Chen, K.M.; Harjes, E.; Gross, P.J.; Fahmy, A.; Lu, Y.; Shindo, K.; Harris, R.S.; Matsuo, H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 2008, 452, 116–119. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Ronquist, F. Bayesian inference of character evolution. Trends Ecol. Evol. 2004, 19, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.J. Markov chain Monte Carlo maximum likelihood. Computing Science and Statistics 1991. In Proceedings of the 23rd Symposium on the Interface, Seattle, WA, USA, 21–24 April 1991; pp. 156–163. [Google Scholar]
- Meyer, X. Adaptive Tree Proposals for Bayesian Phylogenetic Inference. Syst. Biol. 2021, 70, 1015–1032. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Silvestri, G.; Hahn, B.H.; Bibollet-Ruche, F.; Gokcumen, O.; Simon, V.; Ooms, M. Vif proteins from diverse primate lentiviral lineages use the same binding site in APOBEC3G. J. Virol. 2013, 87, 11861–11871. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Geller, R.; Garijo, R.; Lopez-Aldeguer, J.; Sanjuan, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef]
- Quinlan, E.M.; King, J.J.; Amemiya, C.T.; Hsu, E.; Larijani, M. Biochemical Regulatory Features of Activation-Induced Cytidine Deaminase Remain Conserved from Lampreys to Humans. Mol. Cell. Biol. 2017, 37, e00077-17. [Google Scholar] [CrossRef]
- Hix, M.A.; Cisneros, G.A. Computational Investigation of APOBEC3H Substrate Orientation and Selectivity. J. Phys. Chem. B 2020, 124, 3903–3908. [Google Scholar] [CrossRef]
- Vasudevan, A.A.J.; Balakrishnan, K.; Gertzen, C.G.W.; Borvető, F.; Zhang, Z.; Sangwiman, A.; Held, U.; Küstermann, C.; Banerjee, S.; Schumann, G.G.; et al. Loop 1 of APOBEC3C regulates its antiviral activity against HIV-1. bioRxiv 2020, 432, 1–67. [Google Scholar] [CrossRef]
- Mitra, M.; Singer, D.; Mano, Y.; Hritz, J.; Nam, G.; Gorelick, R.J.; Byeon, I.J.; Gronenborn, A.M.; Iwatani, Y.; Levin, J.G. Sequence and structural determinants of human APOBEC3H deaminase and anti-HIV-1 activities. Retrovirology 2015, 12, 3. [Google Scholar] [CrossRef]
- Silvas, T.V.; Schiffer, C.A. APOBEC3s: DNA-editing human cytidine deaminases. Protein Sci. A Publ. Protein Soc. 2019, 28, 1552–1566. [Google Scholar] [CrossRef]
- Ghimire-Rijal, S.; Maynard, E.L., Jr. Comparative thermodynamic analysis of zinc binding to the His/Cys motif in virion infectivity factor. Inorg. Chem. 2014, 53, 4295–4302. [Google Scholar] [CrossRef]
- Smith, J.L.; Izumi, T.; Borbet, T.C.; Hagedorn, A.N.; Pathak, V.K. HIV-1 and HIV-2 Vif interact with human APOBEC3 proteins using completely different determinants. J. Virol. 2014, 88, 9893–9908. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ito, F.; Wolfe, A.D.; Li, S.; Mohammadzadeh, N.; Love, R.P.; Yan, M.; Zirkle, B.; Gaba, A.; Chelico, L.; et al. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat. Commun. 2020, 11, 632. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Myint, W.; Delviks-Frankenberry, K.A.; Hou, S.; Kanai, T.; Balachandran, V.; Sierra Rodriguez, C.; Tripathi, R.; Kurt Yilmaz, N.; Pathak, V.K.; et al. Crystal Structure of a Soluble APOBEC3G Variant Suggests ssDNA to Bind in a Channel that Extends between the Two Domains. J. Mol. Biol. 2020, 432, 6042–6060. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Booiman, T.; Kootstra, N.; Simon, V.; Ooms, M. Identification of the HIV-1 Vif and Human APOBEC3G Protein Interface. Cell Rep. 2015, 13, 1789–1799. [Google Scholar] [CrossRef]
- Zennou, V.; Bieniasz, P.D. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology 2006, 349, 31–40. [Google Scholar] [CrossRef]
- Hayward, J.A.; Tachedjian, M.; Cui, J.; Cheng, A.Z.; Johnson, A.; Baker, M.L.; Harris, R.S.; Wang, L.F.; Tachedjian, G. Differential Evolution of Antiretroviral Restriction Factors in Pteropid Bats as Revealed by APOBEC3 Gene Complexity. Mol. Biol. Evol. 2018, 35, 1626–1637. [Google Scholar] [CrossRef]
- Xiao, X.; Yang, H.; Arutiunian, V.; Fang, Y.; Besse, G.; Morimoto, C.; Zirkle, B.; Chen, X.S. Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res. 2017, 45, 7494–7506. [Google Scholar] [CrossRef]
- Maiti, A.; Hedger, A.K.; Myint, W.; Balachandran, V.; Watts, J.K.; Schiffer, C.A.; Matsuo, H. Structure of the catalytically active APOBEC3G bound to a DNA oligonucleotide inhibitor reveals tetrahedral geometry of the transition state. Nat. Commun. 2022, 13, 7117. [Google Scholar] [CrossRef]
- Li, Y.L.; Langley, C.A.; Azumaya, C.M.; Echeverria, I.; Chesarino, N.M.; Emerman, M.; Cheng, Y.; Gross, J.D. The structural basis for HIV-1 Vif antagonism of human APOBEC3G. Nature 2023, 615, 728–733. [Google Scholar] [CrossRef]
- Shi, K.; Demir, O.; Carpenter, M.A.; Banerjee, S.; Harki, D.A.; Amaro, R.E.; Harris, R.S.; Aihara, H. Active site plasticity and possible modes of chemical inhibition of the human DNA deaminase APOBEC3B. FASEB Bioadv. 2020, 2, 49–58. [Google Scholar] [CrossRef]
- Abdouni, H.S.; King, J.J.; Ghorbani, A.; Fifield, H.; Berghuis, L.; Larijani, M. DNA/RNA hybrid substrates modulate the catalytic activity of purified AID. Mol. Immunol. 2018, 93, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Azimi, F.C.; Lee, J.E. Structural perspectives on HIV-1 Vif and APOBEC3 restriction factor interactions. Protein Sci. A Publ. Protein Soc. 2020, 29, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Webb, D.M. Rapid evolution of primate antiviral enzyme APOBEC3G. Hum. Mol. Genet. 2004, 13, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Etienne, L.; Bibollet-Ruche, F.; Sudmant, P.H.; Wu, L.I.; Hahn, B.H.; Emerman, M. The Role of the Antiviral APOBEC3 Gene Family in Protecting Chimpanzees against Lentiviruses from Monkeys. PLoS Pathog. 2015, 11, e1005149. [Google Scholar] [CrossRef]
- Liu, M.C.; Liao, W.Y.; Buckley, K.M.; Yang, S.Y.; Rast, J.P.; Fugmann, S.D. AID/APOBEC-like cytidine deaminases are ancient innate immune mediators in invertebrates. Nat. Commun. 2018, 9, 1948. [Google Scholar] [CrossRef]
- Krishnan, A.; Iyer, L.M.; Holland, S.J.; Boehm, T.; Aravind, L. Diversification of AID/APOBEC-like deaminases in metazoa: Multiplicity of clades and widespread roles in immunity. Proc. Natl. Acad. Sci. USA 2018, 115, E3201–E3210. [Google Scholar] [CrossRef]
- Basu, U.; Wang, Y.; Alt, F.W. Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Mol. Cell 2008, 32, 285–291. [Google Scholar] [CrossRef]
- Wakae, K.; Magor, B.G.; Saunders, H.; Nagaoka, H.; Kawamura, A.; Kinoshita, K.; Honjo, T.; Muramatsu, M. Evolution of class switch recombination function in fish activation-induced cytidine deaminase, AID. Int. Immunol. 2006, 18, 41–47. [Google Scholar] [CrossRef]
- Chen, Q.; Xiao, X.; Wolfe, A.; Chen, X.S. The in vitro Biochemical Characterization of an HIV-1 Restriction Factor APOBEC3F: Importance of Loop 7 on Both CD1 and CD2 for DNA Binding and Deamination. J. Mol. Biol. 2016, 428, 2661–2670. [Google Scholar] [CrossRef]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef]
- Bransteitter, R.; Prochnow, C.; Chen, X.S. The current structural and functional understanding of APOBEC deaminases. Cell. Mol. Life Sci. 2009, 66, 3137–3147. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Thomas, C.J.; Petersen-Mahrt, S.K.; Neuberger, M.S. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 2005, 22, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Losey, H.C.; Ruthenburg, A.J.; Verdine, G.L. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 2006, 13, 153–159. [Google Scholar] [CrossRef]
- Bitra, A.; Biswas, A.; Anand, R. Structural basis of the substrate specificity of cytidine deaminase superfamily Guanine deaminase. Biochemistry 2013, 52, 8106–8114. [Google Scholar] [CrossRef]
- Jia, Q.; Zhang, J.; Zeng, H.; Tang, J.; Xiao, N.; Gao, S.; Li, H.; Xie, W. Substrate Specificity of GSDA Revealed by Cocrystal Structures and Binding Studies. Int. J. Mol. Sci. 2022, 23, 14976. [Google Scholar] [CrossRef]
- Dolce, L.G.; Zimmer, A.A.; Tengo, L.; Weis, F.; Rubio, M.A.T.; Alfonzo, J.D.; Kowalinski, E. Structural basis for sequence-independent substrate selection by eukaryotic wobble base tRNA deaminase ADAT2/3. Nat. Commun. 2022, 13, 6737. [Google Scholar] [CrossRef]
- Thuy-Boun, A.S.; Thomas, J.M.; Grajo, H.L.; Palumbo, C.M.; Park, S.; Nguyen, L.T.; Fisher, A.J.; Beal, P.A. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. 2020, 48, 7958–7972. [Google Scholar] [CrossRef]
- Minasov, G.; Wawrzak, Z.; Skarina, T.; Wang, Y.; Grimshaw, S.; Papazisi, L.; Savchenko, A.; Anderson, W.F.; Center for Structural Genomics of Infectious Diseases (CSGID). 2.2 Angstrom Crystal Structure of Cytidine deaminase from Vibrio cholerae in Complex with Zinc and Uridine. Protein Data Bank 2012. [Google Scholar] [CrossRef]
- Teh, A.H.; Kimura, M.; Yamamoto, M.; Tanaka, N.; Yamaguchi, I.; Kumasaka, T. The 1.48 A resolution crystal structure of the homotetrameric cytidine deaminase from mouse. Biochemistry 2006, 45, 7825–7833. [Google Scholar] [CrossRef]
- Marx, A.; Alian, A. The first crystal structure of a dTTP-bound deoxycytidylate deaminase validates and details the allosteric-inhibitor binding site. J. Biol. Chem. 2015, 290, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.M.D.; Bohn, M.F.; Kelch, B.A.; Royer, W.E.; Somasundaran, M.; Kurt Yilmaz, N.; Matsuo, H.; et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.L.; Starrett, G.J.; Duffy, J.V.; Demir, O.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Delviks-Frankenberry, K.A.; Wu, C.; Arizaga, F.; Pathak, V.K.; Xiong, Y. Structural insights into PPP2R5A degradation by HIV-1 Vif. Nat. Struct. Mol. Biol. 2024, 31, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Albin, J.S.; Demir, O.; Shaban, N.M.; Luengas, E.M.; Land, A.M.; Anderson, B.D.; Holten, J.R.; Anderson, J.S.; Harki, D.A.; et al. The Binding Interface between Human APOBEC3F and HIV-1 Vif Elucidated by Genetic and Computational Approaches. Cell Rep. 2015, 13, 1781–1788. [Google Scholar] [CrossRef]
- Nakashima, M.; Tsuzuki, S.; Awazu, H.; Hamano, A.; Okada, A.; Ode, H.; Maejima, M.; Hachiya, A.; Yokomaku, Y.; Watanabe, N.; et al. Mapping Region of Human Restriction Factor APOBEC3H Critical for Interaction with HIV-1 Vif. J. Mol. Biol. 2017, 429, 1262–1276. [Google Scholar] [CrossRef]
- Ooms, M.; Letko, M.; Simon, V. The Structural Interface between HIV-1 Vif and Human APOBEC3H. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Smith, J.L.; Pathak, V.K. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J. Virol. 2010, 84, 12599–12608. [Google Scholar] [CrossRef]
- Modenini, G.; Abondio, P.; Boattini, A. The coevolution between APOBEC3 and retrotransposons in primates. Mob. DNA 2022, 13, 27. [Google Scholar] [CrossRef]
- Shaban, N.M.; Yan, R.; Shi, K.; Moraes, S.N.; Cheng, A.Z.; Carpenter, M.A.; McLellan, J.S.; Yu, Z.; Harris, R.S. Cryo-EM structure of the EBV ribonucleotide reductase BORF2 and mechanism of APOBEC3B inhibition. Sci. Adv. 2022, 8, eabm2827. [Google Scholar] [CrossRef]
- Ito, F.; Yang, H.; Xiao, X.; Li, S.X.; Wolfe, A.; Zirkle, B.; Arutiunian, V.; Chen, X.S. Understanding the Structure, Multimerization, Subcellular Localization and mC Selectivity of a Genomic Mutator and Anti-HIV Factor APOBEC3H. Sci. Rep. 2018, 8, 3763. [Google Scholar] [CrossRef]
- Shaban, N.M.; Shi, K.; Lauer, K.V.; Carpenter, M.A.; Richards, C.M.; Salamango, D.; Wang, J.; Lopresti, M.W.; Banerjee, S.; Levin-Klein, R.; et al. The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol. Cell 2018, 69, 75–86 e79. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Demir, O.; Carpenter, M.A.; Wagner, J.; Kurahashi, K.; Harris, R.S.; Amaro, R.E.; Aihara, H. Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci. Rep. 2017, 7, 17415. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huebert, D.N.G.; Ghorbani, A.; Lam, S.Y.B.; Larijani, M. Coevolution of Lentiviral Vif with Host A3F and A3G: Insights from Computational Modelling and Ancestral Sequence Reconstruction. Viruses 2025, 17, 393. https://doi.org/10.3390/v17030393
Huebert DNG, Ghorbani A, Lam SYB, Larijani M. Coevolution of Lentiviral Vif with Host A3F and A3G: Insights from Computational Modelling and Ancestral Sequence Reconstruction. Viruses. 2025; 17(3):393. https://doi.org/10.3390/v17030393
Chicago/Turabian StyleHuebert, David Nicolas Giuseppe, Atefeh Ghorbani, Shaw Yick Brian Lam, and Mani Larijani. 2025. "Coevolution of Lentiviral Vif with Host A3F and A3G: Insights from Computational Modelling and Ancestral Sequence Reconstruction" Viruses 17, no. 3: 393. https://doi.org/10.3390/v17030393
APA StyleHuebert, D. N. G., Ghorbani, A., Lam, S. Y. B., & Larijani, M. (2025). Coevolution of Lentiviral Vif with Host A3F and A3G: Insights from Computational Modelling and Ancestral Sequence Reconstruction. Viruses, 17(3), 393. https://doi.org/10.3390/v17030393