

Evolution of a Distinct SARS-CoV-2 Lineage Identified during an Investigation of a Hospital Outbreak
 ,
,
Abstract
1. Introduction
2. Methods
3. Results
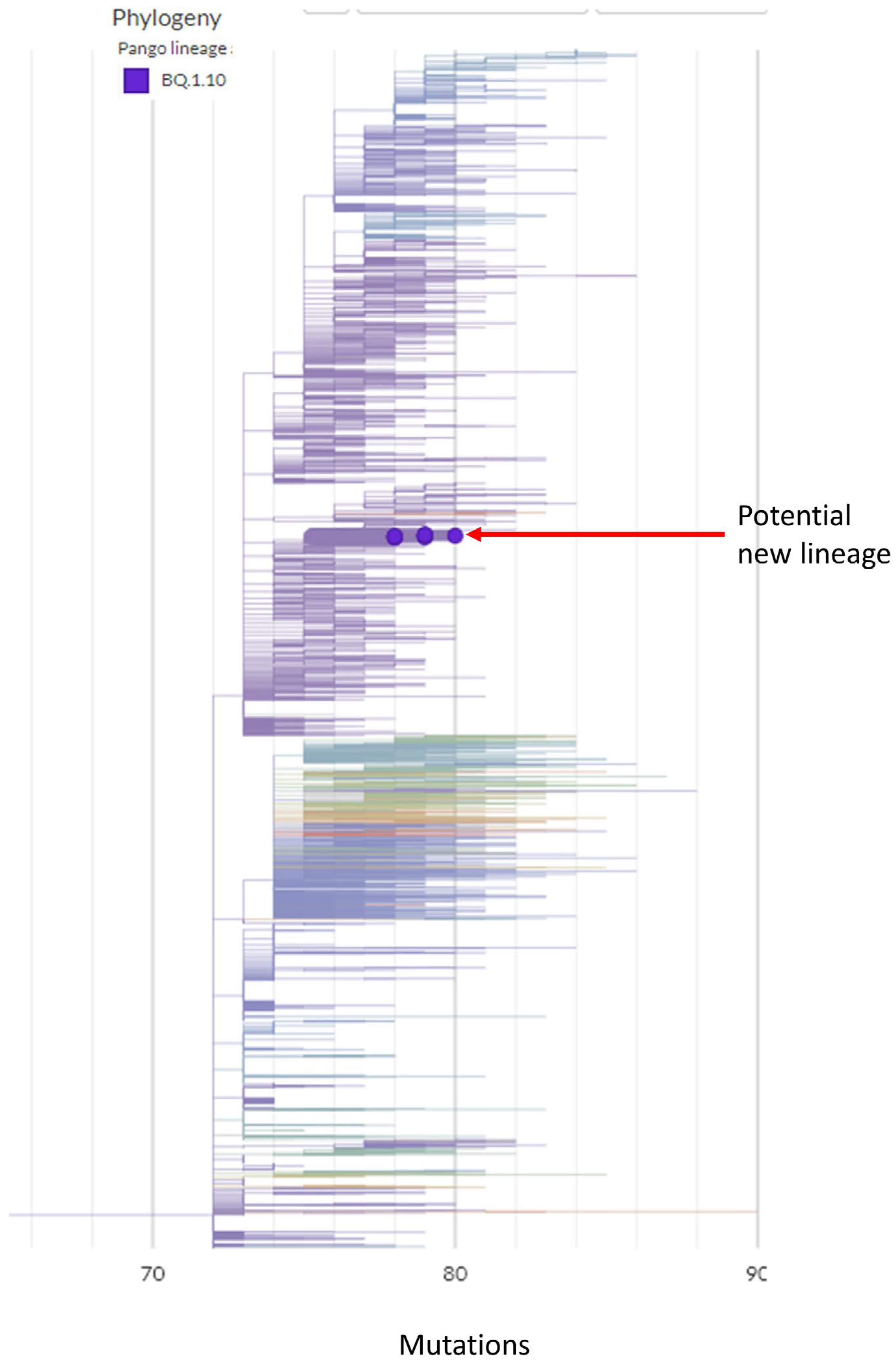
3.1. A Potential New Omicron Sub-Lineage from Hospital Outbreak
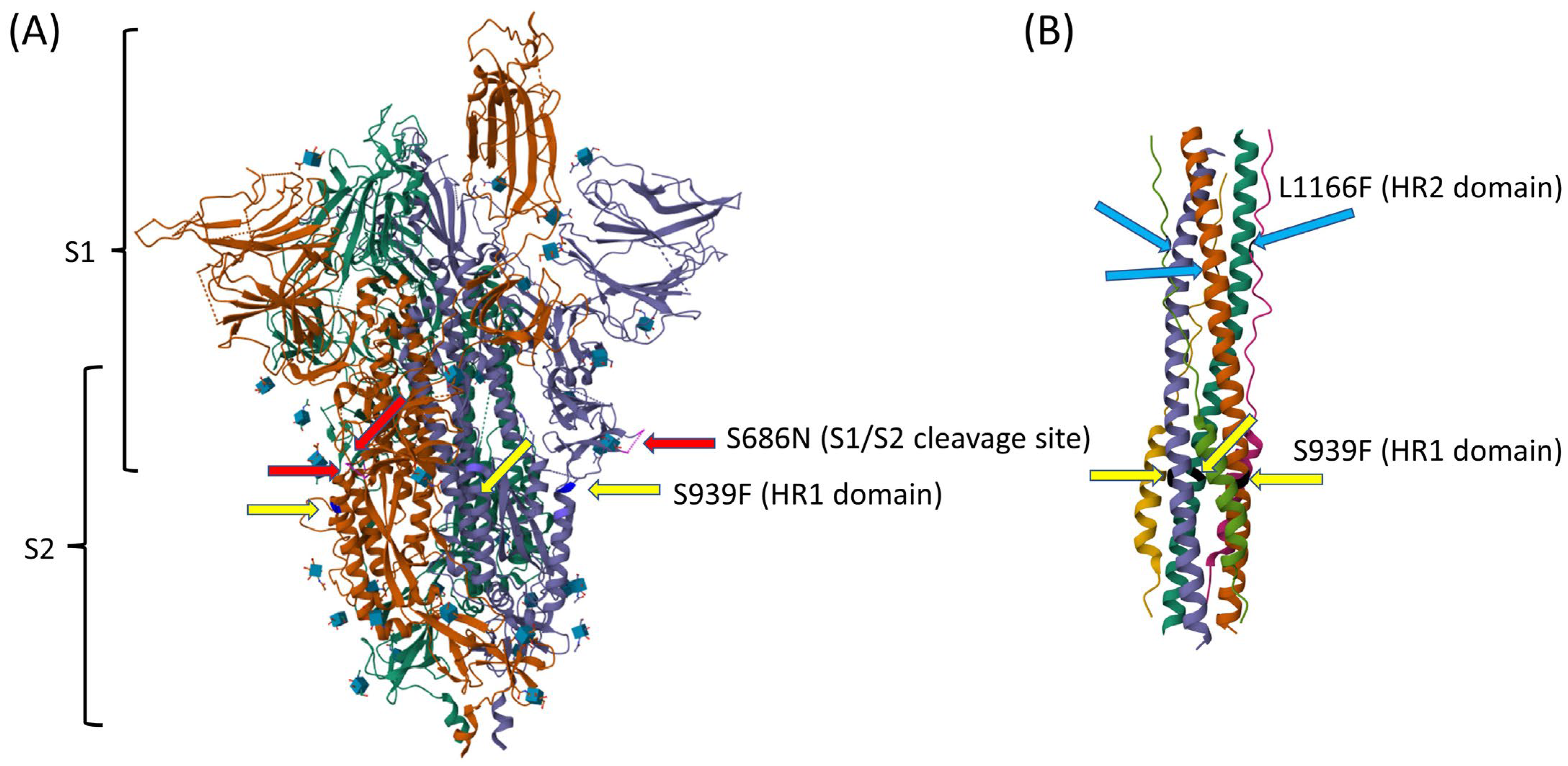
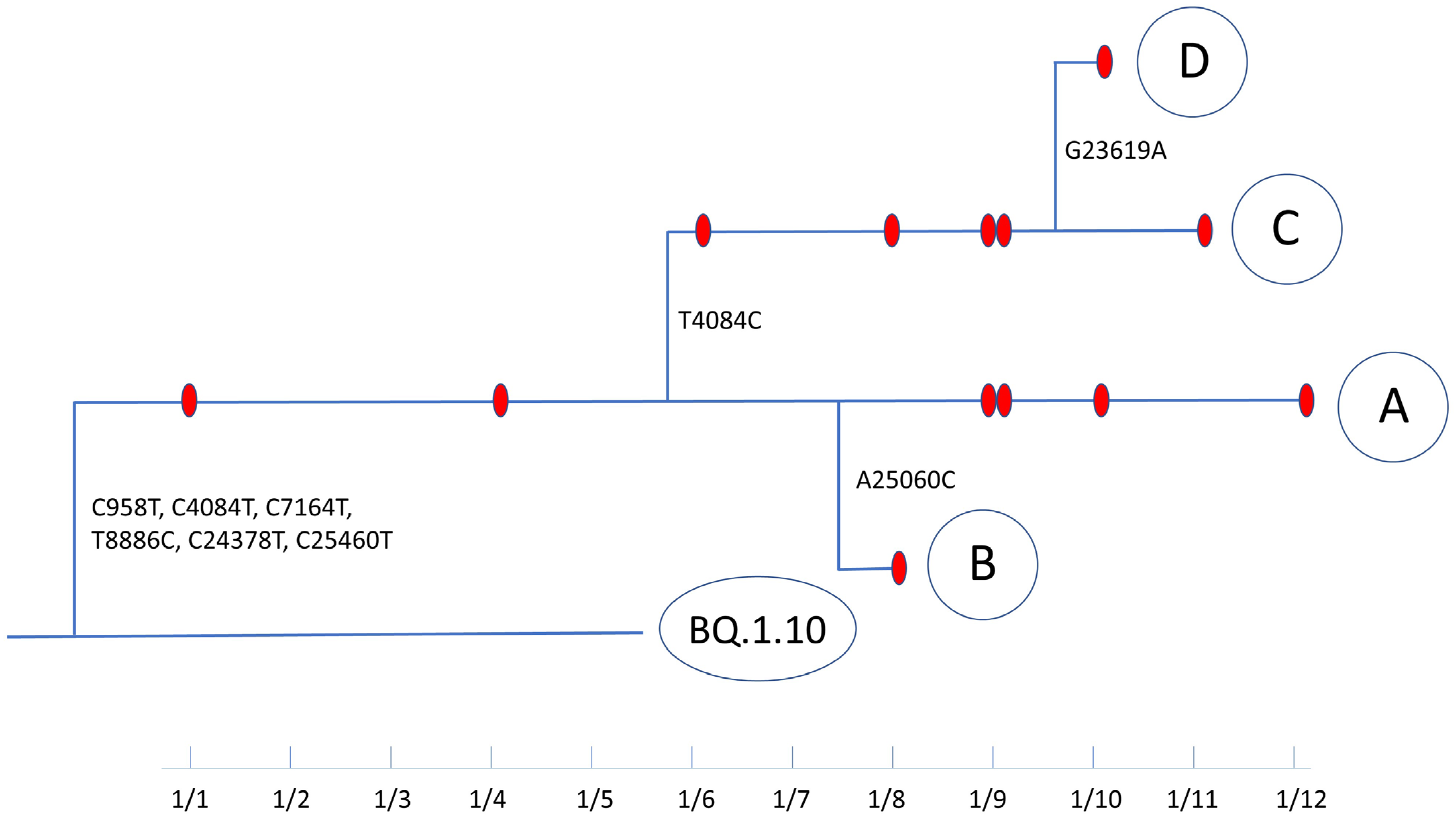
3.2. Evolutionary Path of a Potential New Omicron Sub-Lineage
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Choi, H.; Chatterjee, P.; Hwang, M.; Lichtfouse, E.; Sharma, V.K.; Jinadatha, C. The viral phoenix: Enhanced infectivity and immunity evasion of SARS-CoV-2 variants. Environ. Chem. Lett. 2022, 20, 1539–1544. [Google Scholar] [CrossRef]
- Kumar, A.; Parashar, R.; Kumar, S.; Faiq, M.A.; Kumari, C.; Kulandhasamy, M.; Narayan, R.K.; Jha, R.K.; Singh, H.N.; Prasoon, P.; et al. Emerging SARS-CoV-2 variants can potentially break set epidemiological barriers in COVID-19. J. Med. Virol. 2022, 94, 1300–1314. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Hwang, M.; Navarathna, D.H.; Xu, J.; Lukey, J.; Jinadatha, C. Performance of COVIDSeq and Swift Normalase Amplicon SARS-CoV-2 Panels for SARS-CoV-2 Genome Sequencing: Practical Guide and Combining FASTQ Strategy. J. Clin. Microbiol. 2022, 60, e0002522. [Google Scholar] [CrossRef]
- Donzelli, S.; Spinella, F.; di Domenico, E.G.; Pontone, M.; Cavallo, I.; Orlandi, G.; Iannazzo, S.; Ricciuto, G.M.; Isg Virology Covid, T.; Pellini, R.; et al. Evidence of a SARS-CoV-2 double Spike mutation D614G/S939F potentially affecting immune response of infected subjects. Comput. Struct. Biotechnol. J. 2022, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef]
- Angelini, M.M.; Neuman, B.W.; Buchmeier, M.J. Untangling membrane rearrangement in the nidovirales. DNA Cell Biol. 2014, 33, 122–127. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, Q.; He, X.; Zhao, C.; Ren, W.; Yang, Z.; Wang, Z.; Ding, Q.; Deng, H.; Wang, T.; et al. Loss of Spike N370 glycosylation as an important evolutionary event for the enhanced infectivity of SARS-CoV-2. Cell Res. 2022, 32, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, C.; Kreutzberger, A.J.B.; Ojha, R.; Kuivanen, S.; Couoh-Cardel, S.; Muratcioglu, S.; Eisen, T.J.; White, K.I.; Held, R.G.; et al. Nanomolar inhibition of SARS-CoV-2 infection by an unmodified peptide targeting the prehairpin intermediate of the spike protein. Proc. Natl. Acad. Sci. USA 2022, 119, e2210990119. [Google Scholar] [CrossRef]
- Li, Y.; Hou, F.; Zhou, M.; Yang, X.; Yin, B.; Jiang, W.; Xu, H. C-to-U RNA deamination is the driving force accelerating SARS-CoV-2 evolution. Life Sci. Alliance 2023, 6, e202201688. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2, E275. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Harris, E. CDC Assesses Risk From BA.2.86, Highly Mutated COVID-19 Variant. JAMA 2023, 330, 1029. [Google Scholar] [CrossRef]
- Scarpa, F.; Ciccozzi, M. On the SARS-CoV-2 BA.2.86 lineage: A mutation point of view. J. Med. Virol. 2023, 95, e29079. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G.; Wilson, C.; Panariello, F.; Soares, V.C.; Dias, S.S.G.; et al. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.M.; Ciling, A.; Taha, T.Y.; Chen, I.P.; Khalid, M.M.; Sreekumar, B.; Chen, P.Y.; Kumar, G.R.; Suryawanshi, R.; Silva, I.; et al. Omicron mutations enhance infectivity and reduce antibody neutralization of SARS-CoV-2 virus-like particles. Proc. Natl. Acad. Sci. USA 2022, 119, e2200592119. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D.; et al. Nucleocapsid mutations in SARS-CoV-2 augment replication and pathogenesis. PLoS Pathog. 2022, 18, e1010627. [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.A.; Topolsky, I.; Manceau, M.; Huisman, J.S.; Jablonski, K.P.; Fuhrmann, L.; Dreifuss, D.; Jahn, K.; Beckmann, C.; et al. Quantification of the spread of SARS-CoV-2 variant B.1.1.7 in Switzerland. Epidemics 2021, 37, 100480. [Google Scholar] [CrossRef]
- Jinadatha, C.; Jones, L.D.; Choi, H.; Chatterjee, P.; Hwang, M.; Redmond, S.N.; Navas, M.E.; Zabarsky, T.F.; Bhullar, D.; Cadnum, J.L.; et al. Transmission of SARS-CoV-2 in Inpatient and Outpatient Settings in a Veterans Affairs Health Care System. Open Forum Infect. Dis. 2021, 8, ofab328. [Google Scholar] [CrossRef] [PubMed]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Ze-Ze, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2002, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; Gralinski, L.E.; Johnson, C.E.; Yao, W.; Kovarova, M.; Dinnon, K.H., 3rd; Liu, H.; Madden, V.J.; Krzystek, H.M.; De, C.; et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 2021, 591, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Zhou, L.; Zhou, Y.; Yan, H.; Lan, K. Emerging SARS-CoV-2 variants: Why, how, and what’s next? Cell Insight 2022, 1, 100029. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005, 1, e11. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Miteva, D.; Kitanova, M.; Batselova, H.; Lazova, S.; Chervenkov, L.; Peshevska-Sekulovska, M.; Sekulovski, M.; Gulinac, M.; Vasilev, G.V.; Tomov, L.; et al. The End or a New Era of Development of SARS-CoV-2 Virus: Genetic Variants Responsible for Severe COVID-19 and Clinical Efficacy of the Most Commonly Used Vaccines in Clinical Practice. Vaccines 2023, 11, 1181. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Dong, N.; Chan, E.W.; Chen, S. Genetic cluster analysis of SARS-CoV-2 and the identification of those responsible for the major outbreaks in various countries. Emerg. Microbes Infect. 2020, 9, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | GISAID ID | Collection Date | Unique Mutations +,++ | Ct | Lineage | Sequence Coverage | Coverage Depth |
|---|---|---|---|---|---|---|---|
| A | hCoV-19/USA/TX-DM-9/2023 | 1 January 2023 | C958T, C4084T, C7164T (ORF1a: T2300I) T8886C (ORF1a: L2874S), C24378T (S: S939F), C25460T (ORF3a: A23V) | 28.5 | BQ.1.10 | 96.97% | 1619 |
| hCoV-19/USA/TX-DM-1/2023 | 4 January 2023 | 24.2 | BQ.1.10 | 97.38% | 2000 | ||
| hCoV-19/USA/TX-DM-8/2023 | 9 January 2023 | 27.4 | BQ.1.10 | 98.02% | 1713 | ||
| hCoV-19/USA/TX-DM-5/2023 | 9 January 2023 | 21.5 | BQ.1.10 | 98.45% | 1904 | ||
| hCoV-19/USA/TX-DM-11/2023 | 10 January 2023 | 29.6 | BQ.1.10 | 97.06% | 1546 | ||
| hCoV-19/USA/TX-DM-13/2023 | 12 January 2023 | 23.6 | BQ.1.10 | 97.99% | 1463 | ||
| B | hCoV-19/USA/TX-DM-4/2023 | 8 January 2023 | C958T, C4084T, C7164T (ORF1a: T2300I) T8886C (ORF1a: L2874S), C24378T (S: S939F), A25060C (S: L1166F), C25460T (ORF3a: A23V) | 28.8 | BQ.1.10 | 96.76% | 1283 |
| C | hCoV-19/USA/TX-DM-2/2023 | 16 January 2023 | C958T, T4084C, C7164T (ORF1a: T2300I) T8886C (ORF1a: L2874S), C24378T (S: S939F), C25460T (ORF3a: A23V) | 20.8 | BQ.1.10 | 98.42% | 2267 |
| hCoV-19/USA/TX-DM-3/2023 | 8 January 2023 | 18.2 | BQ.1.10 | 98.89% | 2075 | ||
| hCoV-19/USA/TX-DM-7/2023 | 9 January 2023 | 17.7 | BQ.1.10 | 99.11% | 2098 | ||
| hCoV-19/USA/TX-DM-6/2023 | 9 January 2023 | 21.5 | BQ.1.10 | 98.80% | 1890 | ||
| hCoV-19/USA/TX-DM-12/2023 | 11 January 2023 | 17.3 | BQ.1.10 | 98.99% | 2566 | ||
| D | hCoV-19/USA/TX-DM-10/2023 | 10 January 2023 | C958T, T4084C, C7164T (ORF1a: T2300I) T8886C (ORF1a: L2874S), G23619A (S: S686N), C24378T (S: S939F), C25460T (ORF3a: A23V) | 22.8 | BQ.1.10 | 98.50% | 1962 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Hwang, M.; Cornelius, L.; Navarathna, D.H.; Chatterjee, P.; Jinadatha, C. Evolution of a Distinct SARS-CoV-2 Lineage Identified during an Investigation of a Hospital Outbreak. Viruses 2024, 16, 337. https://doi.org/10.3390/v16030337
Choi H, Hwang M, Cornelius L, Navarathna DH, Chatterjee P, Jinadatha C. Evolution of a Distinct SARS-CoV-2 Lineage Identified during an Investigation of a Hospital Outbreak. Viruses. 2024; 16(3):337. https://doi.org/10.3390/v16030337
Chicago/Turabian StyleChoi, Hosoon, Munok Hwang, Lisa Cornelius, Dhammika H. Navarathna, Piyali Chatterjee, and Chetan Jinadatha. 2024. "Evolution of a Distinct SARS-CoV-2 Lineage Identified during an Investigation of a Hospital Outbreak" Viruses 16, no. 3: 337. https://doi.org/10.3390/v16030337
APA StyleChoi, H., Hwang, M., Cornelius, L., Navarathna, D. H., Chatterjee, P., & Jinadatha, C. (2024). Evolution of a Distinct SARS-CoV-2 Lineage Identified during an Investigation of a Hospital Outbreak. Viruses, 16(3), 337. https://doi.org/10.3390/v16030337