Targeting Viral and Cellular Cysteine Proteases for Treatment of New Variants of SARS-CoV-2


 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Materials
2.3. SARS-CoV-2 3CLpro Pro and PLpro Luminescent Assays
2.4. Viability Assay
2.5. Production of SARS-CoV-2 Pseudotyped Particles
2.6. In Vitro Pseudovirus Entry Inhibition Assay
2.7. Molecular Docking
2.8. Molecular Dynamics Simulations
2.9. Statistical Analysis
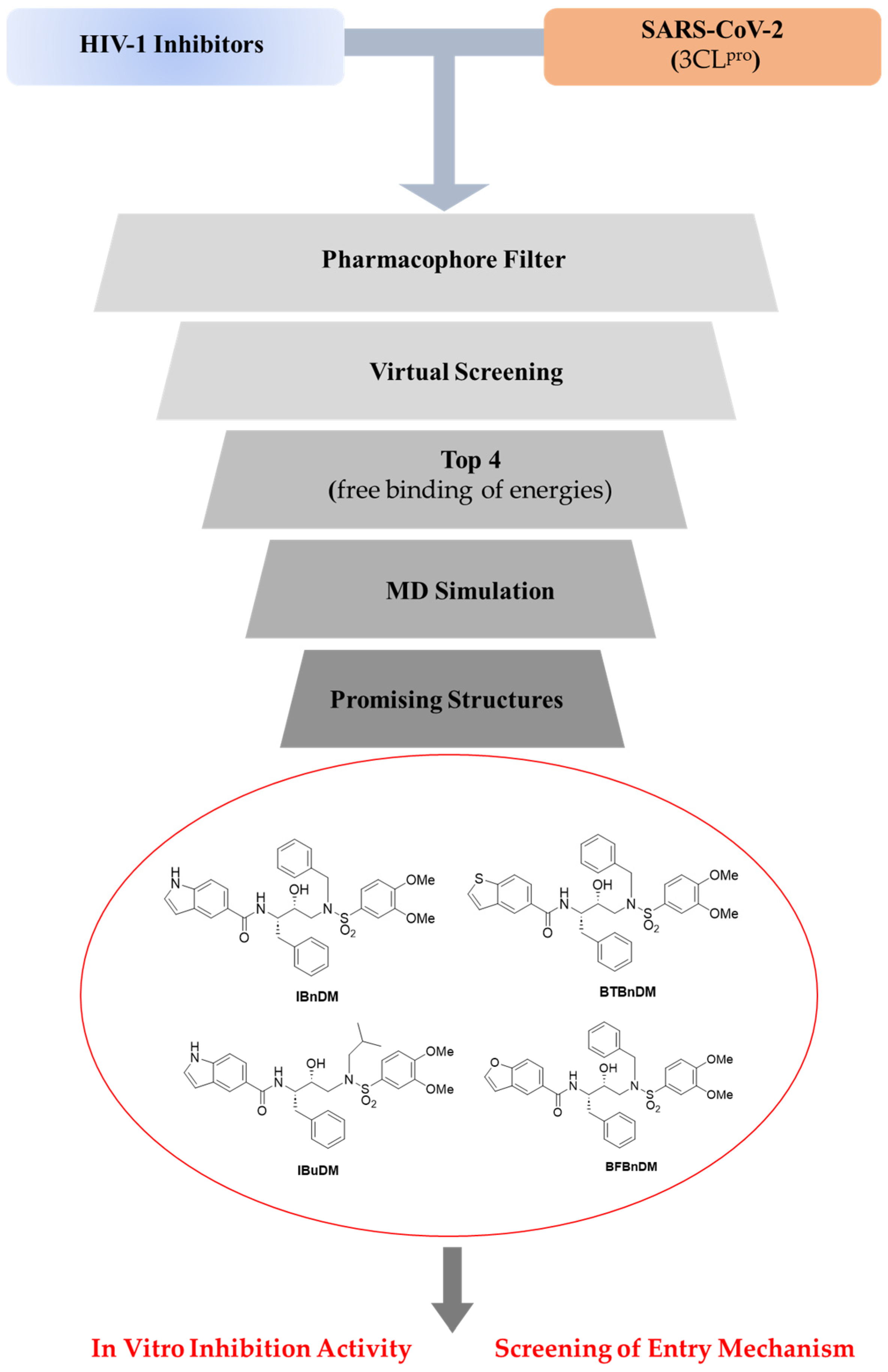
3. Results
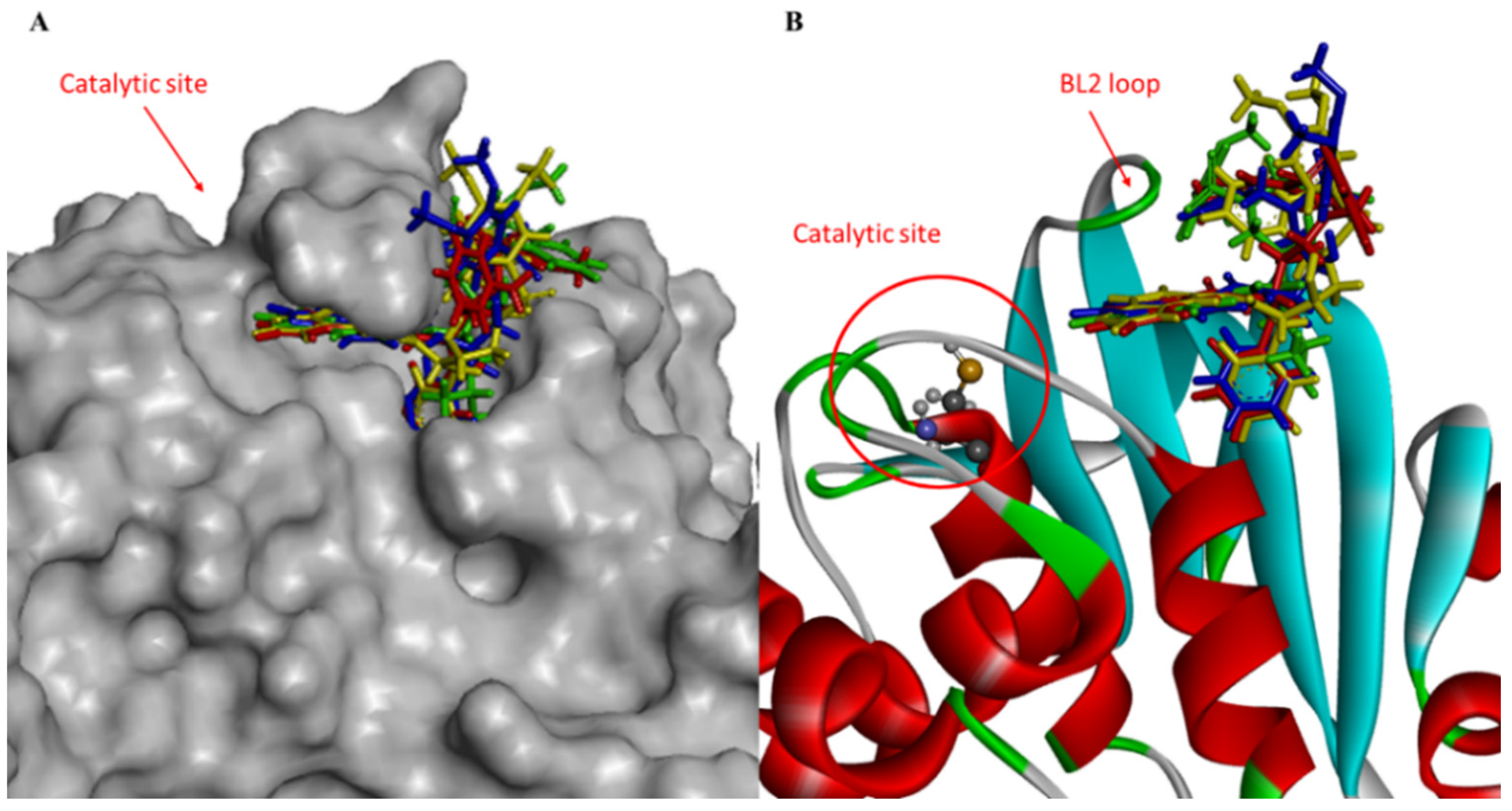
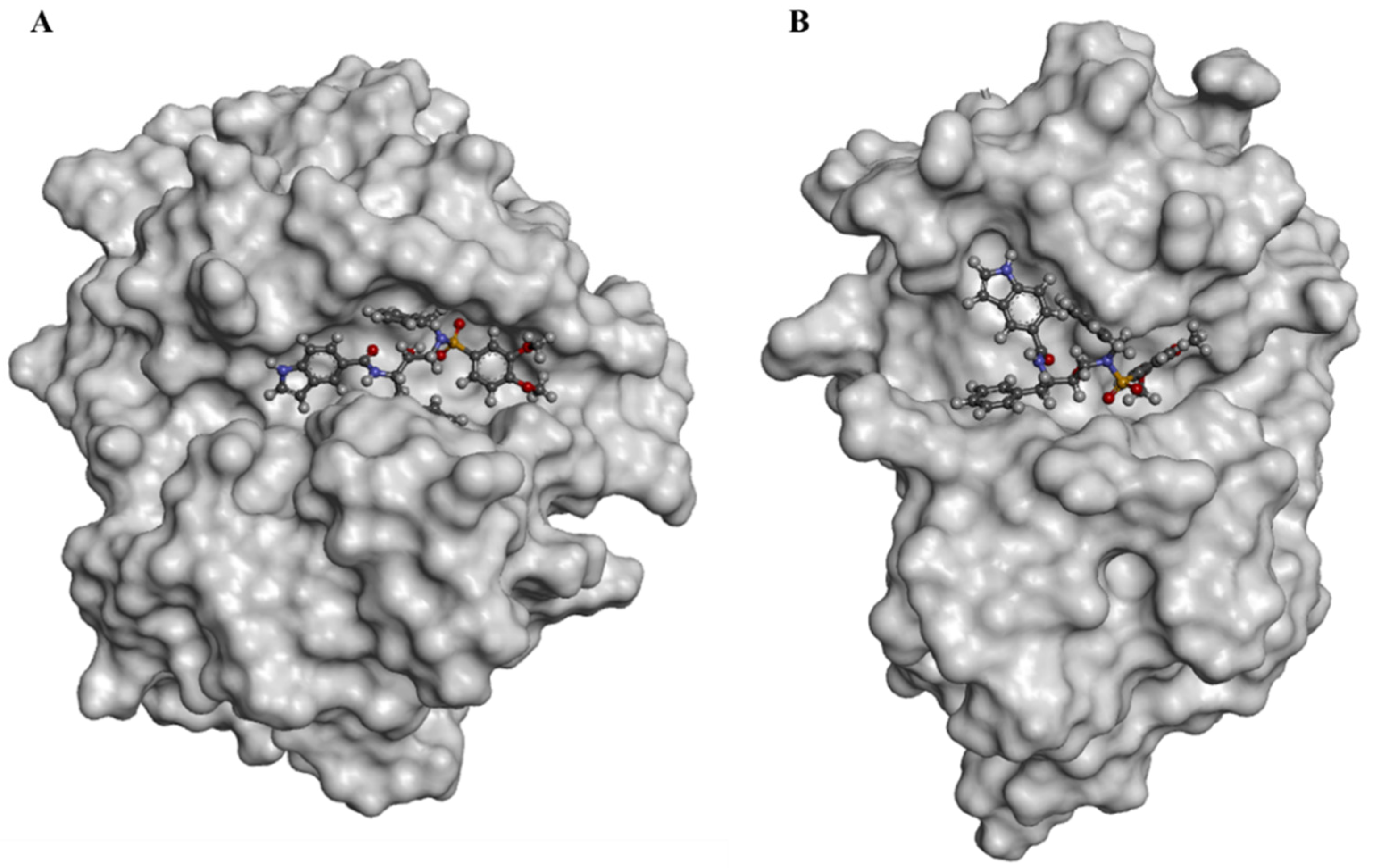
3.1. Docking and Molecular Dynamics Simulations
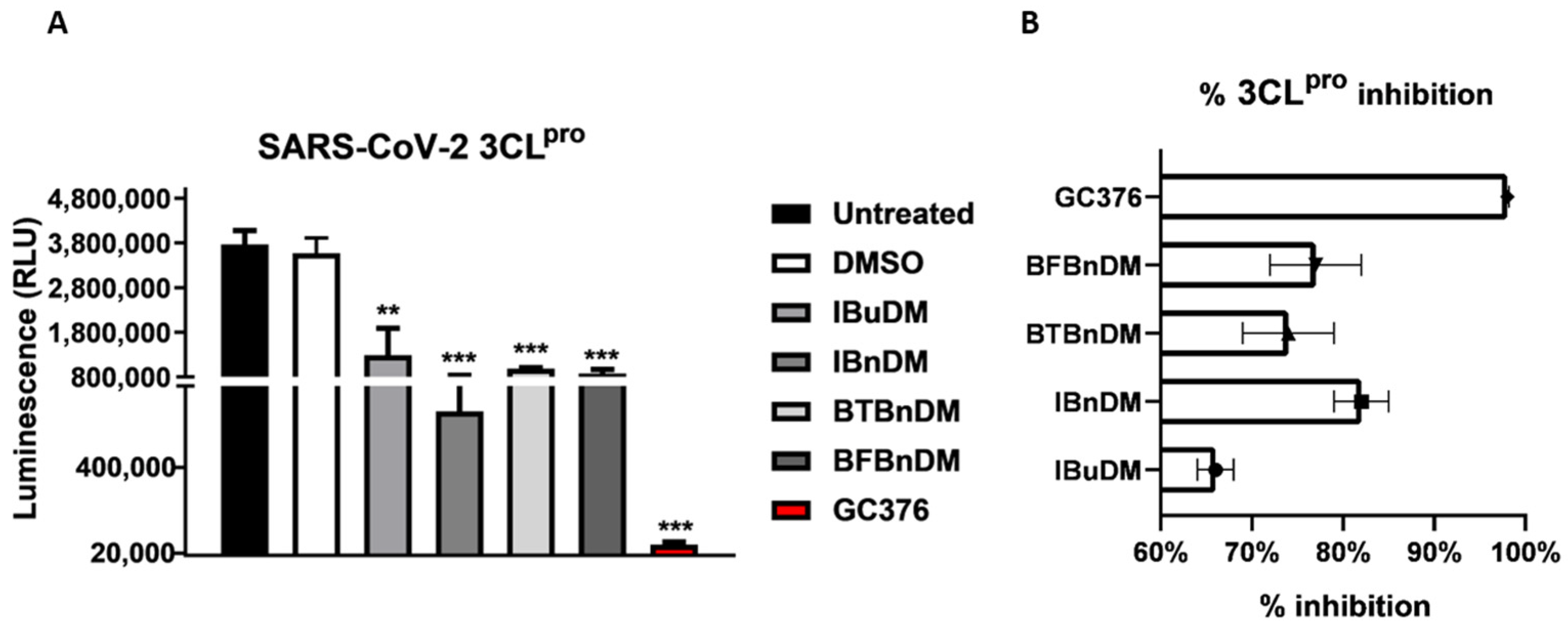
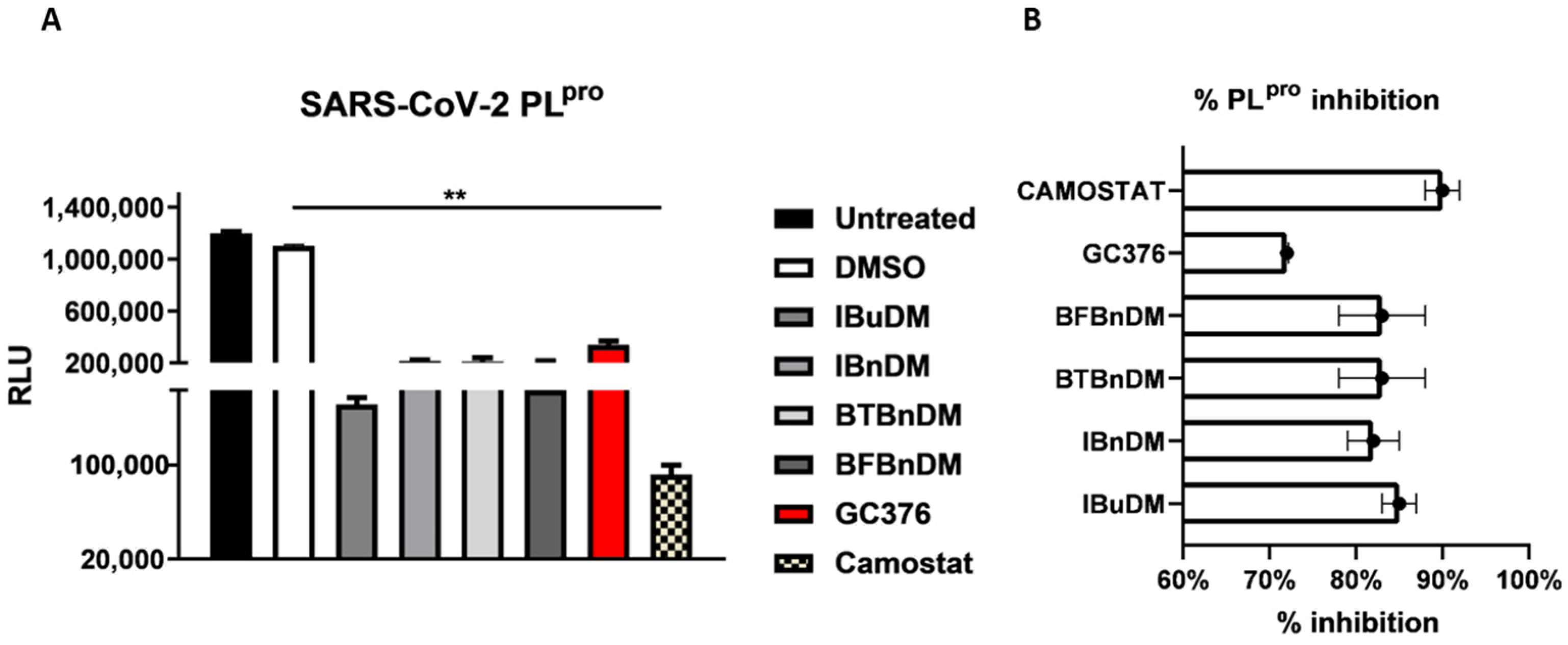
3.2. In-Vitro Screening for 3CLpro and PLpro Inhibition
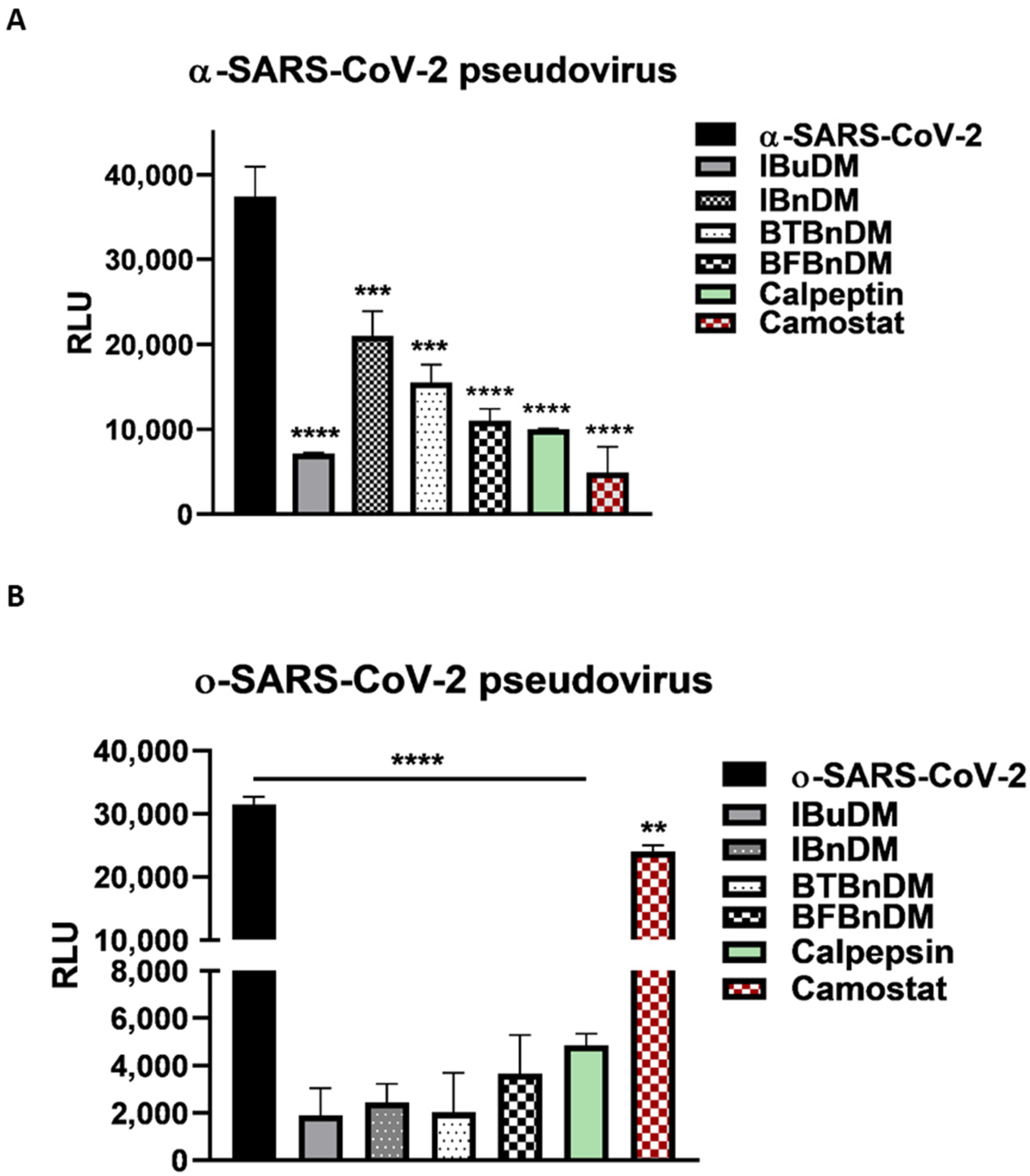
3.3. Inhibition of SARS-CoV-2 Spike Pseudotyped Particle Transduction by Using Cathepsin and TMPRSS2 Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease Targeted COVID-19 Drug Discovery and Its Challenges: Insight into Viral Main Protease (Mpro) and Papain-like Protease (PLpro) Inhibitors. Bioorg. Med. Chem. 2021, 29, 115860. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tőzsér, J. Analysis of the Efficacy of HIV Protease Inhibitors against SARS-CoV-2′s Main Protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef]
- Chen, F.; Chan, K.H.; Jiang, Y.; Kao, R.Y.T.; Lu, H.T.; Fan, K.W.; Cheng, V.C.C.; Tsui, W.H.W.; Hung, I.F.N.; Lee, T.S.W. In Vitro Susceptibility of 10 Clinical Isolates of SARS Coronavirus to Selected Antiviral Compounds. J. Clin. Virol. 2004, 31, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative Therapeutic Efficacy of Remdesivir and Combination Lopinavir, Ritonavir, and Interferon Beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of Potential Molecules against COVID-19 Main Protease through Structure-Guided Virtual Screening Approach. J. Biomol. Struct. Dyn. 2021, 39, 3662–3680. [Google Scholar] [CrossRef]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M. Role of Lopinavir/Ritonavir in the Treatment of SARS: Initial Virological and Clinical Findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.-T.; Wong, A.Y.-L.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.-H.; Huang, X.; et al. Remdesivir, Lopinavir, Emetine, and Homoharringtonine Inhibit SARS-CoV-2 Replication in Vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Dimasi, A.; Moi, D.; Passarella, D.; Scala, A.; Piperno, A.; Micale, N. Recent Advances in SARS-CoV-2 Main Protease Inhibitors: From Nirmatrelvir to Future Perspectives. Biomolecules 2023, 13, 1339. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against Mpro and Cathepsin L. Sci. Adv. 2020, 6, abe0751. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct Conformational States of SARS-CoV-2 Spike Protein. Science (1979) 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Cell Editorial Team. Embracing the Landscape of Therapeutics. Cell 2020, 181, 1–3. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Pöhlmann, S. Priming Time: How Cellular Proteases Arm Coronavirus Spike Proteins. In Activation of Viruses by Host Proteases; Springer International Publishing: Cham, Swizerland, 2018; pp. 71–98. [Google Scholar]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Xiang, Q.; Li, L.; Wu, J.; Tian, M.; Fu, Y. Application of Pseudovirus System in the Development of Vaccine, Antiviral-Drugs, and Neutralizing Antibodies. Microbiol. Res. 2022, 258, 126993. [Google Scholar] [CrossRef] [PubMed]
- Salazar-García, M.; Acosta-Contreras, S.; Rodríguez-Martínez, G.; Cruz-Rangel, A.; Flores-Alanis, A.; Patiño-López, G.; Luna-Pineda, V.M. Pseudotyped Vesicular Stomatitis Virus-Severe Acute Respiratory Syndrome-Coronavirus-2 Spike for the Study of Variants, Vaccines, and Therapeutics Against Coronavirus Disease 2019. Front. Microbiol. 2022, 12, 817200. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive Exploration of Chemical Space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed]
- Funicello, M.; Chiummiento, L.; Tramutola, F.; Armentano, M.F.; Bisaccia, F.; Miglionico, R.; Milella, L.; Benedetti, F.; Berti, F.; Lupattelli, P. Synthesis and Biological Evaluation in Vitro and in Mammalian Cells of New Heteroaryl Carboxyamides as HIV-Protease Inhibitors. Bioorg. Med. Chem. 2017, 25, 4715–4722. [Google Scholar] [CrossRef] [PubMed]
- D’Orsi, R.; Funicello, M.; Laurita, T.; Lupattelli, P.; Berti, F.; Chiummiento, L. The Pseudo-Symmetric N-Benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modeling and Biological Evaluation. Biomolecules 2021, 11, 1584. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.; Whittaker, G. Murine Leukemia Virus (MLV)-Based Coronavirus Spike-Pseudotyped Particle Production and Infection. Bio Protoc. 2016, 6, e2035. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Tang, T.; Nathan, L.; Jaimes, J.A.; Hsu, H.-L.; Daniel, S.; Whittaker, G.R. Production of Pseudotyped Particles to Study Highly Pathogenic Coronaviruses in a Biosafety Level 2 Setting. J. Vis. Exp. 2019, 145, e59010. [Google Scholar] [CrossRef]
- Gentile, D.; Floresta, G.; Patamia, V.; Nicosia, A.; Mineo, P.G.; Rescifina, A. Cucurbit[7]Uril as a Catalytic Nanoreactor for One-Pot Synthesis of Isoxazolidines in Water. Org. Biomol. Chem. 2020, 18, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Floresta, G.; Patamia, V.; Chiaramonte, R.; Mauro, G.L.; Rescifina, A.; Vecchio, M. An Integrated Pharmacophore/Docking/3D-QSAR Approach to Screening a Large Library of Products in Search of Future Botulinum Neurotoxin a Inhibitors. Int. J. Mol. Sci. 2020, 21, 9470. [Google Scholar] [CrossRef]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-Approved Drugs for Treating Iatrogenic Botulism: A Paired 3D-QSAR/Docking Approach. ChemMedChem 2020, 15, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat Mesylate Inhibits SARS-CoV-2 Activation by TMPRSS2-Related Proteases and Its Metabolite GBPA Exerts Antiviral Activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, R.; Gentile, D.; Rescifina, A.; Napoli, E.; Trischitta, P.; Piperno, A.; Sciortino, M.T. An Integrated In Silico and In Vitro Approach for the Identification of Natural Products Active against SARS-CoV-2. Biomolecules 2023, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Chitsike, L.; Duerksen-Hughes, P. Keep out! SARS-CoV-2 Entry Inhibitors: Their Role and Utility as COVID-19 Therapeutics. Virol. J. 2021, 18, 154. [Google Scholar] [CrossRef]
- Metzdorf, K.; Jacobsen, H.; Greweling-Pils, M.C.; Hoffmann, M.; Lüddecke, T.; Miller, F.; Melcher, L.; Kempf, A.M.; Nehlmeier, I.; Bruder, D.; et al. TMPRSS2 Is Essential for SARS-CoV-2 Beta and Omicron Infection. Viruses 2023, 15, 271. [Google Scholar] [CrossRef]
- Kutkat, O.; Moatasim, Y.; Al-Karmalawy, A.A.; Abulkhair, H.S.; Gomaa, M.R.; El-Taweel, A.N.; Abo Shama, N.M.; GabAllah, M.; Mahmoud, D.B.; Kayali, G.; et al. Robust Antiviral Activity of Commonly Prescribed Antidepressants against Emerging Coronaviruses: In Vitro and in Silico Drug Repurposing Studies. Sci. Rep. 2022, 12, 12920. [Google Scholar] [CrossRef]
- Lewnard, J.A.; McLaughlin, J.M.; Malden, D.; Hong, V.; Puzniak, L.; Ackerson, B.K.; Lewin, B.J.; Kim, J.S.; Shaw, S.F.; Takhar, H.; et al. Effectiveness of Nirmatrelvir–Ritonavir in Preventing Hospital Admissions and Deaths in People with COVID-19: A Cohort Study in a Large US Health-Care System. Lancet Infect. Dis. 2023, 23, 806–815. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H.; Kwon, Y.; Li, Y.; Alqarni, S.; Huang, F.; Dubrovskyi, O.; et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [Google Scholar] [CrossRef] [PubMed]
- Saroha, B.; Kumar, G.; Kumari, M.; Kaur, R.; Raghav, N.; Sharma, P.K.; Kumar, N.; Kumar, S. A Decennary Update on Diverse Heterocycles and Their Intermediates as Privileged Scaffolds for Cathepsin B Inhibition. Int. J. Biol. Macromol. 2022, 222, 2270–2308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Zhu, Y.; Zhang, L.; Zhong, G.; Tai, L.; Liu, S.; Yin, G.; Lu, J.; He, Q.; Li, M.-J.; et al. Novel Cleavage Sites Identified in SARS-CoV-2 Spike Protein Reveal Mechanism for Cathepsin L-Facilitated Viral Infection and Treatment Strategies. Cell Discov. 2022, 8, 53. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, D.; Chiummiento, L.; Santarsiere, A.; Funicello, M.; Lupattelli, P.; Rescifina, A.; Venuti, A.; Piperno, A.; Sciortino, M.T.; Pennisi, R. Targeting Viral and Cellular Cysteine Proteases for Treatment of New Variants of SARS-CoV-2. Viruses 2024, 16, 338. https://doi.org/10.3390/v16030338
Gentile D, Chiummiento L, Santarsiere A, Funicello M, Lupattelli P, Rescifina A, Venuti A, Piperno A, Sciortino MT, Pennisi R. Targeting Viral and Cellular Cysteine Proteases for Treatment of New Variants of SARS-CoV-2. Viruses. 2024; 16(3):338. https://doi.org/10.3390/v16030338
Chicago/Turabian StyleGentile, Davide, Lucia Chiummiento, Alessandro Santarsiere, Maria Funicello, Paolo Lupattelli, Antonio Rescifina, Assunta Venuti, Anna Piperno, Maria Teresa Sciortino, and Rosamaria Pennisi. 2024. "Targeting Viral and Cellular Cysteine Proteases for Treatment of New Variants of SARS-CoV-2" Viruses 16, no. 3: 338. https://doi.org/10.3390/v16030338
APA StyleGentile, D., Chiummiento, L., Santarsiere, A., Funicello, M., Lupattelli, P., Rescifina, A., Venuti, A., Piperno, A., Sciortino, M. T., & Pennisi, R. (2024). Targeting Viral and Cellular Cysteine Proteases for Treatment of New Variants of SARS-CoV-2. Viruses, 16(3), 338. https://doi.org/10.3390/v16030338