
Functional Analysis of KAP1/TRIM28 Requirements for HIV-1 Transcription Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Generation of the HCT116:KAP1dTAG Cell Line
2.3. Generation of the HCT116:KAP1dTAG Cell Lines Stably Expressing GFP and KAP1
2.4. Cloning of KAP1 Deletion Constructs
2.5. Site-Directed Mutagenesis
2.6. Luciferase Reporter Assays
2.7. Immunofluorescence and Confocal Microscopy
2.8. Western Blots
3. Results
3.1. Acute KAP1 Depletion Partially Decreases Signal-Induced HIV-1 Reporter Activation
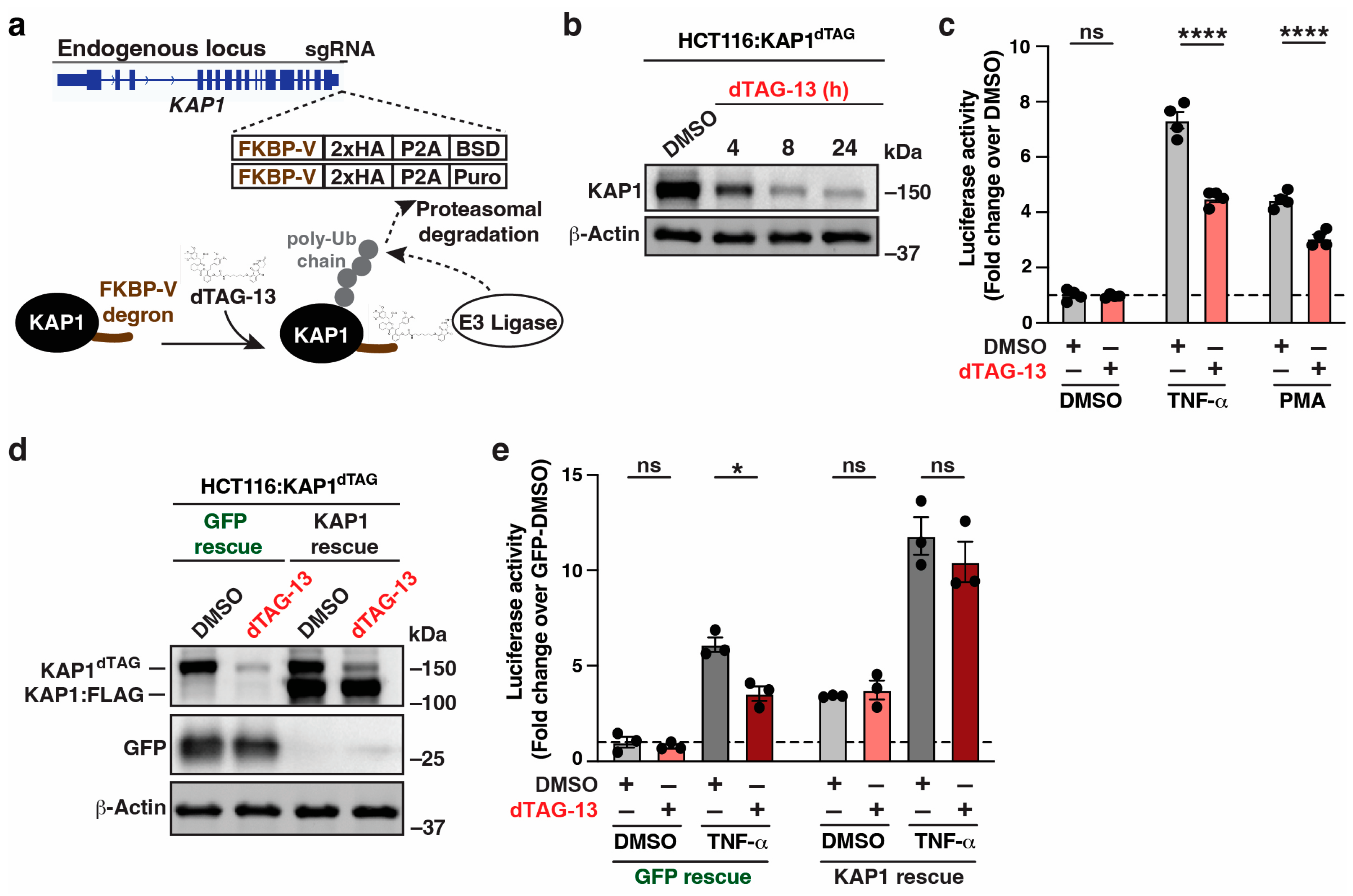
3.2. KAP1 Deletion Analysis Predicts Domains Important for HIV-1 Reporter Activation
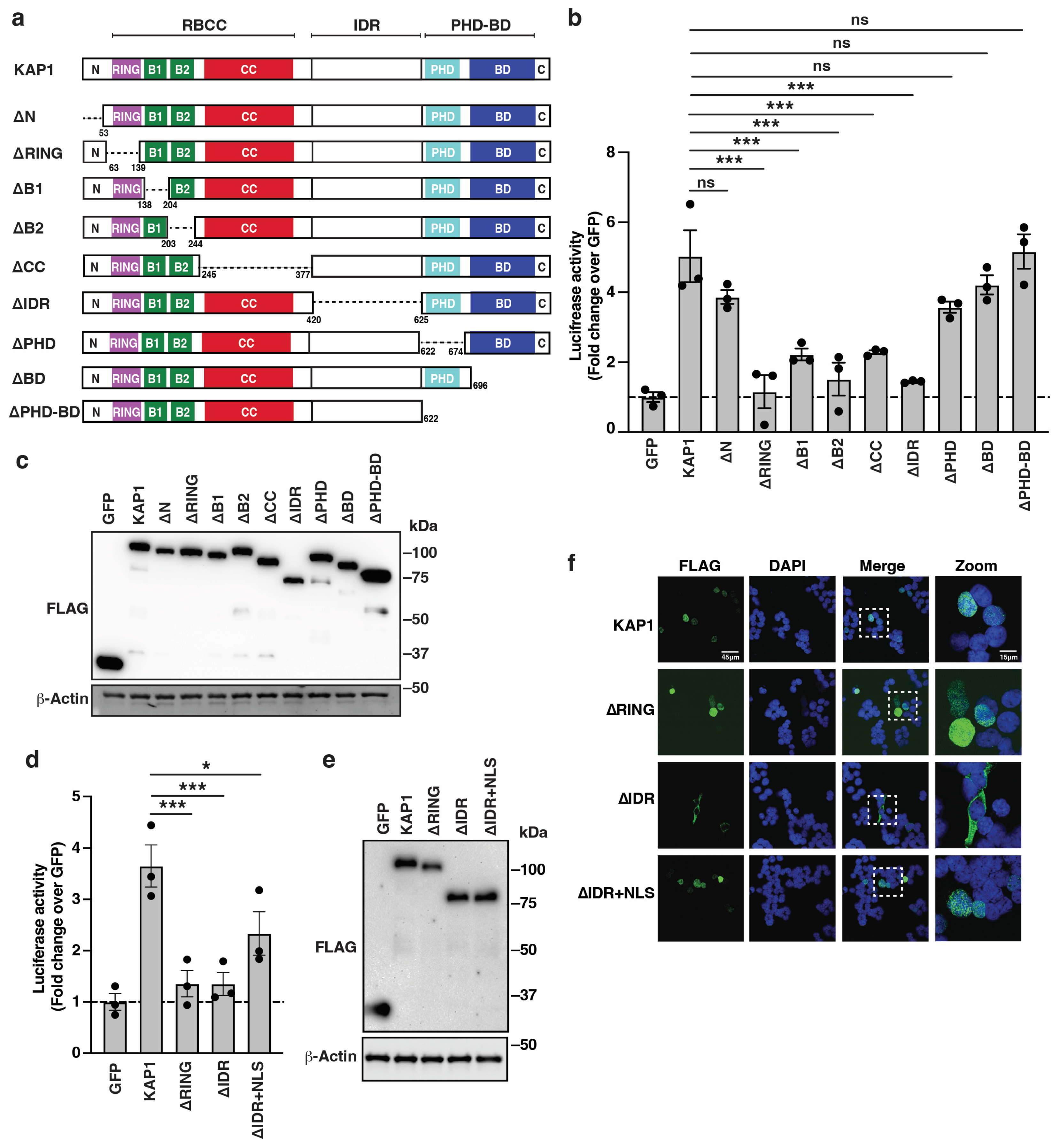
3.3. KAP1 RING Finger, Nuclear Localization, and IDR Are Critical Determinants for HIV-1 Reporter Activation

3.4. The RING E3 Ligase Is Potentially Required in the Nucleus for a General, but Not Pathway-Specific, Transcription Activation Function

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, R.T.; Abreu, C.M.; Costa, P.A.G.; Ferreira, E.A.; Ratliff, J.; Pohlenz, L.; Shirk, E.N.; Rubin, L.H.; Blankson, J.N.; Gama, L.; et al. Monocyte-derived macrophages contain persistent latent HIV reservoirs. Nat. Microbiol. 2023, 8, 833–844. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef]
- Shukla, A.; Ramirez, N.P.; D’Orso, I. HIV-1 Proviral Transcription and Latency in the New Era. Viruses 2020, 12, 555. [Google Scholar] [CrossRef]
- Einkauf, K.B.; Osborn, M.R.; Gao, C.; Sun, W.; Sun, X.; Lian, X.; Parsons, E.M.; Gladkov, G.T.; Seiger, K.W.; Blackmer, J.E.; et al. Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell 2022, 185, 266–282. [Google Scholar] [CrossRef]
- Fauci, A.S.; Redfield, R.R.; Sigounas, G.; Weahkee, M.D.; Giroir, B.P. Ending the HIV Epidemic: A Plan for the United States. JAMA 2019, 321, 844–845. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Arlen, P.A.; Gao, L.; Kitchen, C.M.; Zack, J.A. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc. Natl. Acad. Sci. USA 2003, 100, 12955–12960. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Nayak, S.; Iwasa, J.; Taatjes, D.J. The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell. Biol. 2022, 23, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.B.; Baltimore, D.; Frankel, A.D. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc Natl Acad Sci USA 1991, 88, 4045–4049. [Google Scholar] [CrossRef]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef]
- Nisole, S.; Stoye, J.P.; Saib, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef]
- White, D.; Rafalska-Metcalf, I.U.; Ivanov, A.V.; Corsinotti, A.; Peng, H.; Lee, S.C.; Trono, D.; Janicki, S.M.; Rauscher, F.J., 3rd. The ATM substrate KAP1 controls DNA repair in heterochromatin: Regulation by HP1 proteins and serine 473/824 phosphorylation. Mol. Cancer Res. 2012, 10, 401–414. [Google Scholar] [CrossRef]
- Jang, S.M.; Kauzlaric, A.; Quivy, J.P.; Pontis, J.; Rauwel, B.; Coluccio, A.; Offner, S.; Duc, J.; Turelli, P.; Almouzni, G.; et al. KAP1 facilitates reinstatement of heterochromatin after DNA replication. Nucleic Acids. Res. 2018, 46, 8788–8802. [Google Scholar] [CrossRef]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jonsson, M.E.; Manne, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Brattas, P.L.; Jonsson, M.E.; Fasching, L.; Nelander Wahlestedt, J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 Controls a Gene Regulatory Network Based on Endogenous Retroviruses in Human Neural Progenitor Cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Asimi, V.; Sampath Kumar, A.; Niskanen, H.; Riemenschneider, C.; Hetzel, S.; Naderi, J.; Fasching, N.; Popitsch, N.; Du, M.; Kretzmer, H.; et al. Hijacking of transcriptional condensates by endogenous retroviruses. Nat. Genet. 2022, 54, 1238–1247. [Google Scholar] [CrossRef]
- McNamara, R.P.; Reeder, J.E.; McMillan, E.A.; Bacon, C.W.; McCann, J.L.; D’Orso, I. KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol. Cell 2016, 61, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.P.; Planelles, V.; D’Orso, I. Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell Rep. 2019, 27, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Pfleger, C.; Lai, J.F.; Roan, F.; Sun, S.C.; Ziegler, S.F. KAP1 Regulates Regulatory T Cell Function and Proliferation in Both Foxp3-Dependent and -Independent Manners. Cell Rep. 2018, 23, 796–807. [Google Scholar] [CrossRef]
- Dalgaard, K.; Landgraf, K.; Heyne, S.; Lempradl, A.; Longinotto, J.; Gossens, K.; Ruf, M.; Orthofer, M.; Strogantsev, R.; Selvaraj, M.; et al. Trim28 Haploinsufficiency Triggers Bi-stable Epigenetic Obesity. Cell 2016, 164, 353–364. [Google Scholar] [CrossRef]
- Bacon, C.W.; Challa, A.; Hyder, U.; Shukla, A.; Borkar, A.N.; Bayo, J.; Liu, J.; Wu, S.Y.; Chiang, C.-M.; Kutateladze, T.G.; et al. KAP1 Is a Chromatin Reader that Couples Steps of RNA Polymerase II Transcription to Sustain Oncogenic Programs. Mol. Cell 2020, 78, 1133–1151. [Google Scholar] [CrossRef]
- Randolph, K.; Hyder, U.; D’Orso, I. KAP1/TRIM28: Transcriptional Activator and/or Repressor of Viral and Cellular Programs? Front. Cell. Infect. Microbiol. 2022, 12, 834636. [Google Scholar] [CrossRef] [PubMed]
- Allouch, A.; Di Primio, C.; Alpi, E.; Lusic, M.; Arosio, D.; Giacca, M.; Cereseto, A. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host. Microbe 2011, 9, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. Elife 2019, 8, e42426. [Google Scholar] [CrossRef] [PubMed]
- Taura, M.; Song, E.; Ho, Y.C.; Iwasaki, A. Apobec3A maintains HIV-1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc. Natl. Acad. Sci. USA 2019, 116, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Ren, X.; Kerppola, T.K. KAP1 represses differentiation-inducible genes in embryonic stem cells through cooperative binding with PRC1 and derepresses pluripotency-associated genes. Mol. Cell. Biol. 2014, 34, 2075–2091. [Google Scholar] [CrossRef]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19, e45000. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Jaeger, M.G.; Winter, G.E. Fast-acting chemical tools to delineate causality in transcriptional control. Mol. Cell 2021, 81, 1617–1630. [Google Scholar] [CrossRef]
- Hyder, U.; Challa, A.; Thronton, M.; Nandu, T.; Kraus, W.L.; D’Orso, I. KAP1 negatively regulates elongation kinetics to activate signal-induced transcription. Mendeley Data, 2023; under review. [Google Scholar] [CrossRef]
- D’Orso, I.; Grunwell, J.R.; Nakamura, R.L.; Das, C.; Frankel, A.D. Targeting tat inhibitors in the assembly of human immunodeficiency virus type 1 transcription complexes. J. Virol. 2008, 82, 9492–9504. [Google Scholar] [CrossRef]
- Nissen, R.M.; Yamamoto, K.R. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000, 14, 2314–2329. [Google Scholar] [CrossRef] [PubMed]
- Gioia, L.; Siddique, A.; Head, S.R.; Salomon, D.R.; Su, A.I. A genome-wide survey of mutations in the Jurkat cell line. BMC Genom. 2018, 19, 334. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.M.; Gao, J.; Wang, J.; Yang, M.; Potts, P.R. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 2010, 39, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Begg, G.E.; Schultz, D.C.; Friedman, J.R.; Jensen, D.E.; Speicher, D.W.; Rauscher, F.J., 3rd. Reconstitution of the KRAB-KAP-1 repressor complex: A model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 2000, 295, 1139–1162. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.A.; Oda, S.I.; Chong, Z.S.; Yu, M.; McLaughlin, S.H.; Modis, Y. Structure of KAP1 tripartite motif identifies molecular interfaces required for retroelement silencing. Proc. Natl. Acad. Sci. USA 2019, 116, 15042–15051. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Sangel, P.; Yamaguchi, H.; Obuse, C.; Miyamoto, Y.; Oka, M.; Yoneda, Y. Identification and characterization of a nuclear localization signal of TRIM28 that overlaps with the HP1 box. Biochem. Biophys. Res. Commun. 2015, 462, 201–207. [Google Scholar] [CrossRef]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carre, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.E7. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Ha, B.; Altay, G.; Greenbaum, J.A.; McVicker, G.; et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 2018, 175, 1701–1715.E16. [Google Scholar] [CrossRef]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Thiru, A.; Nietlispach, D.; Mott, H.R.; Okuwaki, M.; Lyon, D.; Nielsen, P.R.; Hirshberg, M.; Verreault, A.; Murzina, N.V.; Laue, E.D. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 2004, 23, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Fonti, G.; Marcaida, M.J.; Bryan, L.C.; Trager, S.; Kalantzi, A.S.; Helleboid, P.J.; Demurtas, D.; Tully, M.D.; Grudinin, S.; Trono, D.; et al. KAP1 is an antiparallel dimer with a functional asymmetry. Life Sci. Alliance 2019, 2, e201900349. [Google Scholar] [CrossRef]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell. Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef]
- Watanabe, M.; Saeki, Y.; Takahashi, H.; Ohtake, F.; Yoshida, Y.; Kasuga, Y.; Kondo, T.; Yaguchi, H.; Suzuki, M.; Ishida, H.; et al. A substrate-trapping strategy to find E3 ubiquitin ligase substrates identifies Parkin and TRIM28 targets. Commun. Biol. 2020, 3, 592. [Google Scholar] [CrossRef]
- Buratowski, S.; Hahn, S.; Guarente, L.; Sharp, P.A. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 1989, 56, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Sawadogo, M.; Roeder, R.G. Factors involved in specific transcription by human RNA polymerase II: Analysis by a rapid and quantitative in vitro assay. Proc. Natl. Acad. Sci. USA 1985, 82, 4394–4398. [Google Scholar] [CrossRef]
- Zhang, Z.; English, B.P.; Grimm, J.B.; Kazane, S.A.; Hu, W.; Tsai, A.; Inouye, C.; You, C.; Piehler, J.; Schultz, P.G.; et al. Rapid dynamics of general transcription factor TFIIB binding during preinitiation complex assembly revealed by single-molecule analysis. Genes Dev. 2016, 30, 2106–2118. [Google Scholar] [CrossRef]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef]
- Wolf, D.; Cammas, F.; Losson, R.; Goff, S.P. Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J. Virol. 2008, 82, 4675–4679. [Google Scholar] [CrossRef]
- Lee, A.; CingOz, O.; Sabo, Y.; Goff, S.P. Characterization of interaction between Trim28 and YY1 in silencing proviral DNA of Moloney murine leukemia virus. Virology 2018, 516, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Yan, J.; Wang, S.; Guo, Y.; Xi, X.; Han, S.; Yin, J.; Peng, B.; He, X.; Bodem, J.; et al. Trim28 acts as restriction factor of prototype foamy virus replication by modulating H3K9me3 marks and destabilizing the viral transactivator Tas. Retrovirology 2021, 18, 38. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Randolph, K.; Hyder, U.; Challa, A.; Perez, E.; D’Orso, I. Functional Analysis of KAP1/TRIM28 Requirements for HIV-1 Transcription Activation. Viruses 2024, 16, 116. https://doi.org/10.3390/v16010116
Randolph K, Hyder U, Challa A, Perez E, D’Orso I. Functional Analysis of KAP1/TRIM28 Requirements for HIV-1 Transcription Activation. Viruses. 2024; 16(1):116. https://doi.org/10.3390/v16010116
Chicago/Turabian StyleRandolph, Keyera, Usman Hyder, Ashwini Challa, Erick Perez, and Iván D’Orso. 2024. "Functional Analysis of KAP1/TRIM28 Requirements for HIV-1 Transcription Activation" Viruses 16, no. 1: 116. https://doi.org/10.3390/v16010116
APA StyleRandolph, K., Hyder, U., Challa, A., Perez, E., & D’Orso, I. (2024). Functional Analysis of KAP1/TRIM28 Requirements for HIV-1 Transcription Activation. Viruses, 16(1), 116. https://doi.org/10.3390/v16010116