
New Concepts in Therapeutic Manipulation of HIV-1 Transcription and Latency: Latency Reversal versus Latency Prevention

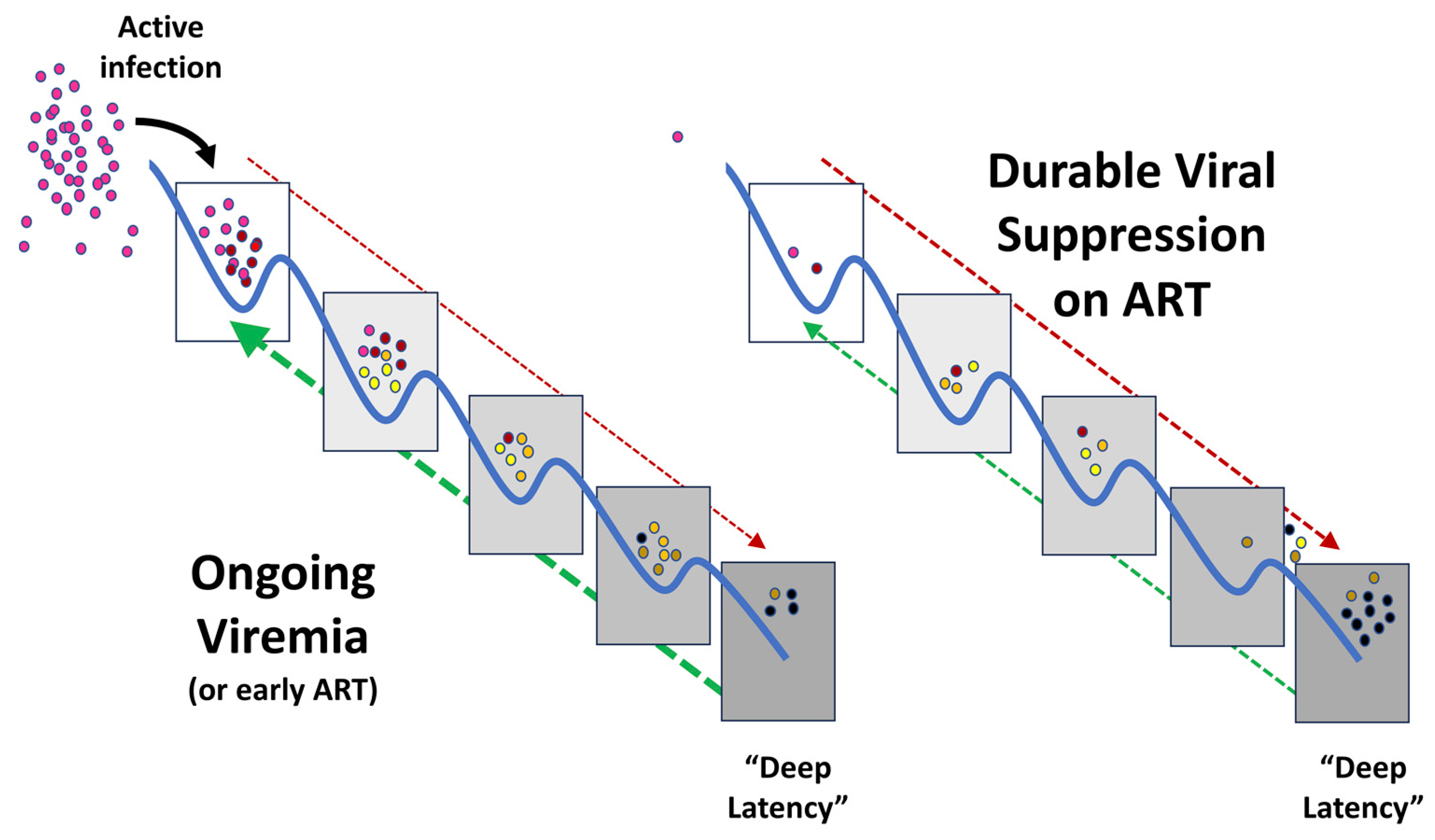{kind=link}
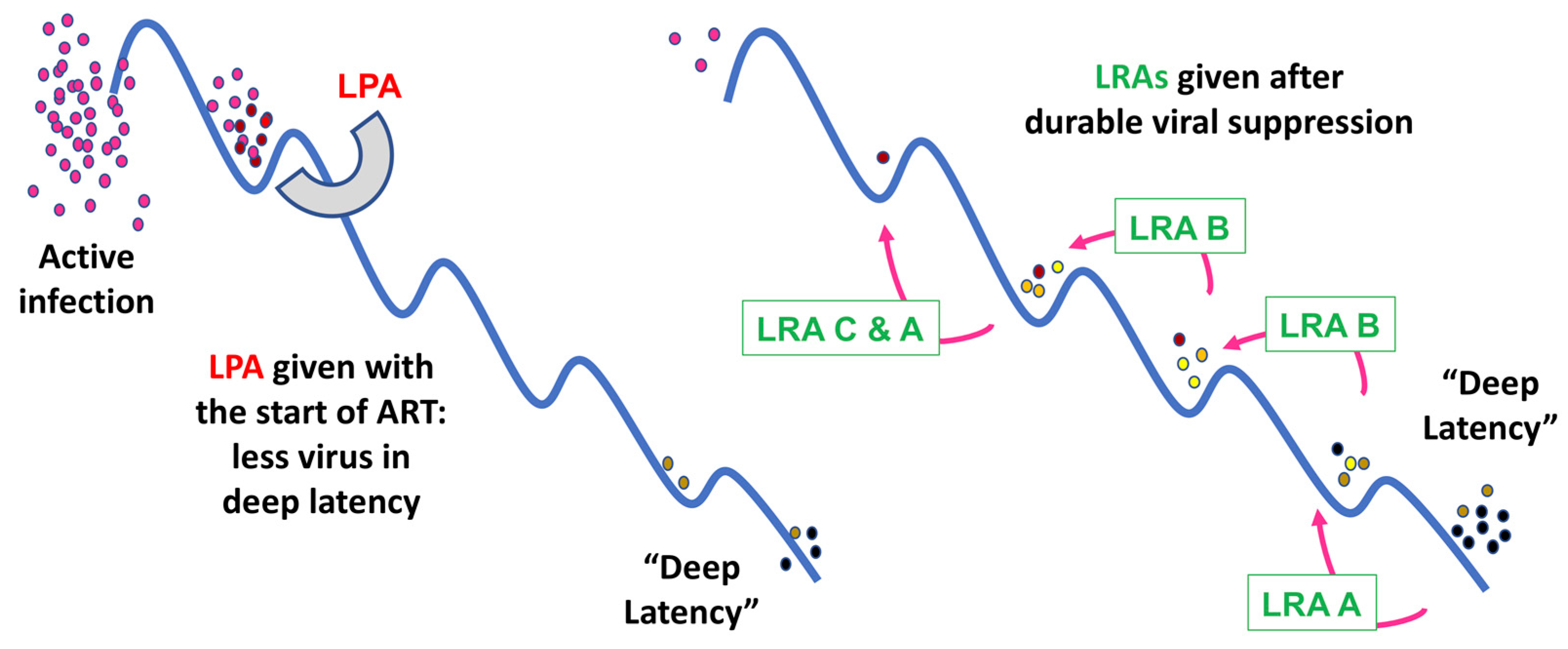{kind=link}
Abstract
1. Introduction
2. Viral Latency: Where and How the Virus Persists
3. Establishment of the Latent Reservoir
4. Molecular Mechanisms of Latency
5. Strategies for Eradicating Latent HIV Reservoirs: Latency Reversing Agents
6. Latency Prevention Agents: A New Concept
7. Therapeutic Challenges
8. Summary/Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Summary of the Global HIV Epidemic. 2020. Available online: https://www.who.int/data/gho/data/themes/hiv-aids (accessed on 4 April 2023).
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Adams, S.V.; Fromentin, R.; Roche, M.; Fling, S.P.; Gonçalves, P.H.; Lurain, K.; Ramaswami, R.; Wang, C.-C.J.; Gorelick, R.J.; et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci. Transl. Med. 2022, 14, eabl3836. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Li, Y.; Hong, J.; Zhang, L. The Rational Combination Strategy of Immunomodulatory Latency Reversing Agents and Novel Immunotherapy to Achieve HIV-1 Cure. Infect. Dis. Immun. 2022, 2, 263. [Google Scholar] [CrossRef]
- Brodin, J.; Zanini, F.; Thebo, L.; Lanz, C.; Bratt, G.; A Neher, R.; Albert, J. Establishment and stability of the latent HIV-1 DNA reservoir. elife 2016, 5, e18889. [Google Scholar] [CrossRef]
- Abrahams, M.-R.; Joseph, S.B.; Garrett, N.; Tyers, L.; Moeser, M.; Archin, N.; Council, O.D.; Matten, D.; Zhou, S.; Doolabh, D.; et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019, 11, eaaw5589. [Google Scholar] [CrossRef]
- Pankau, M.; Reeves, D.B.; Harkins, E.; Ronen, K.; Jaoko, W.; Mandaliya, K.; Graham, S.M.; McClelland, R.S.; Iv, F.A.M.; Schiffer, J.T.; et al. Dynamics of HIV DNA reservoir seeding in a cohort of superinfected Kenyan women. PLoS Pathog. 2020, 16, e1008286. [Google Scholar] [CrossRef]
- Chun, T.-W.; Engel, D.; Mizell, S.B.; Ehler, L.A.; Fauci, A.S. Induction of HIV-1 Replication in Latently Infected CD4+ T Cells Using a Combination of Cytokines. J. Exp. Med. 1998, 188, 83–91. [Google Scholar] [CrossRef]
- Chun, T.-W.; Justement, J.S.; Murray, D.; Hallahan, C.W.; Maenza, J.; Collier, A.C.; Sheth, P.M.; Kaul, R.; Ostrowski, M.; Moir, S.; et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: Implications for eradication. Aids 2010, 24, 2803–2808. [Google Scholar] [CrossRef]
- Chun, T.-W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.M.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Genet. 2016, 14, 55–60. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Crooks, A.M.; Bateson, R.; Cope, A.B.; Dahl, N.P.; Griggs, M.K.; Kuruc, J.D.; Gay, C.L.; Eron, J.J.; Margolis, D.M.; Bosch, R.J.; et al. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. J. Infect. Dis. 2015, 212, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Hiener, B.; Horsburgh, B.A.; Eden, J.-S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.A.; Chun, T.-W.; Justement, S.J.; Motola, I.; Spinelli, M.A.; Adelsberger, J.; Ehler, L.A.; Mizell, S.B.; Hallahan, C.W.; Fauci, A.S. Both Memory and CD45RA +/CD62L + Naive CD4+ T Cells Are Infected in Human Immunodeficiency Virus Type 1-Infected Individuals. J. Virol. 1999, 73, 6430–6435. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Sarabia, N.; Bateson, R.E.; Dahl, N.P.; Crooks, A.M.; Kuruc, J.D.; Margolis, D.M.; Archin, N.M. Quantitation of Replication-Competent HIV-1 in Populations of Resting CD4+ T Cells. J. Virol. 2014, 88, 14070–14077. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell–like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Zerbato, J.M.; Serrao, E.; Lenzi, G.; Kim, B.; Ambrose, Z.; Watkins, S.C.; Engelman, A.N.; Sluis-Cremer, N. Establishment and Reversal of HIV-1 Latency in Naive and Central Memory CD4+ T Cells In Vitro. J. Virol. 2016, 90, 8059–8073. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Chomont, N. HIV persistence in subsets of CD4+ T cells: 50 shades of reservoirs. Semin. Immunol. 2021, 51, 101438. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef]
- Veenhuis, R.T.; Abreu, C.M.; Costa, P.A.G.; Ferreira, E.A.; Ratliff, J.; Pohlenz, L.; Shirk, E.N.; Rubin, L.H.; Blankson, J.N.; Gama, L.; et al. Monocyte-derived macrophages contain persistent latent HIV reservoirs. Nat. Microbiol. 2023, 8, 833–844. [Google Scholar] [CrossRef]
- Tang, Y.; Chaillon, A.; Gianella, S.; Wong, L.M.; Li, D.; Simermeyer, T.L.; Porrachia, M.; Ignacio, C.; Woodworth, B.; Zhong, D.; et al. Brain microglia serve as a persistent HIV reservoir despite durable antiretroviral therapy. J. Clin. Investig. 2023, 133, e167417. [Google Scholar] [CrossRef]
- Anderson, J.L.; Khoury, G.; Fromentin, R.; Solomon, A.; Chomont, N.; Sinclair, E.; Milush, J.M.; Hartogensis, W.; Bacchetti, P.; Roche, M.; et al. Human Immunodeficiency Virus (HIV)–Infected CCR6+ Rectal CD4+ T Cells and HIV Persistence On Antiretroviral Therapy. J. Infect. Dis. 2020, 221, 744–755. [Google Scholar] [CrossRef]
- Gosselin, A.; Salinas, T.R.W.; Planas, D.; Wacleche, V.S.; Zhang, Y.; Fromentin, R.; Chomont, N.; Cohen, A.; Shacklett, B.; Mehraj, V.; et al. HIV persists in CCR6 + CD4+ T cells from colon and blood during antiretroviral therapy. Aids 2017, 31, 35–48. [Google Scholar] [CrossRef]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.-K. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J. Neuroimmune Pharmacol. 2019, 14, 110–119. [Google Scholar] [CrossRef]
- Yukl, S.A.; Gianella, S.; Sinclair, E.; Epling, L.; Li, Q.; Duan, L.; Choi, A.L.M.; Girling, V.; Ho, T.; Li, P.; et al. Differences in HIV Burden and Immune Activation within the Gut of HIV-Positive Patients Receiving Suppressive Antiretroviral Therapy. J. Infect. Dis. 2010, 202, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in Gut-Associated Lymphoid Tissue despite Long-Term Antiretroviral Therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Bosque, A.; Famiglietti, M.; Weyrich, A.S.; Goulston, C.; Planelles, V. Homeostatic Proliferation Fails to Efficiently Reactivate HIV-1 Latently Infected Central Memory CD4+ T Cells. PLoS Pathog. 2011, 7, e1002288. [Google Scholar] [CrossRef]
- Simonetti, F.R.; Zhang, H.; Soroosh, G.P.; Duan, J.; Rhodehouse, K.; Hill, A.L.; Beg, S.A.; McCormick, K.; Raymond, H.E.; Nobles, C.L.; et al. Antigen-driven clonal selection shapes the persistence of HIV-1–infected CD4+ T cells in vivo. J. Clin. Investig. 2021, 131, e145254. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Bale, M.J.; Wells, D.; Guo, S.; Luke, B.; Zerbato, J.M.; Sobolewski, M.D.; Sia, T.; Shao, W.; Wu, X.; et al. Integration in oncogenes plays only a minor role in determining the in vivo distribution of HIV integration sites before or during suppressive antiretroviral therapy. PLoS Pathog. 2021, 17, e1009141. [Google Scholar] [CrossRef]
- Pan, X.; Baldauf, H.-M.; Keppler, O.T.; Fackler, O.T. Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell Res. 2013, 23, 876–885. [Google Scholar] [CrossRef]
- Pierson, T.C.; Zhou, Y.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Siliciano, R.F. Molecular Characterization of Preintegration Latency in Human Immunodeficiency Virus Type 1 Infection. J. Virol. 2002, 76, 8518–8531. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T Lymphocytes as an Inducible Virus Reservoir in HIV-1 Infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef]
- A Zack, J.; Haislip, A.M.; Krogstad, P.; Chen, I.S. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J. Virol. 1992, 66, 1717–1725. [Google Scholar] [CrossRef]
- Soto, P.C.; Terry, V.H.; Lewinski, M.K.; Deshmukh, S.; Beliakova-Bethell, N.; Spina, C.A. HIV-1 latency is established preferentially in minimally activated and non-dividing cells during productive infection of primary CD4 T cells. PLoS ONE 2022, 17, e0271674. [Google Scholar] [CrossRef]
- Luzuriaga, K.; Gay, H.; Ziemniak, C.; Sanborn, K.B.; Somasundaran, M.; Rainwater-Lovett, K.; Mellors, J.W.; Rosenbloom, D.; Persaud, D. Viremic Relapse after HIV-1 Remission in a Perinatally Infected Child. N. Engl. J. Med. 2015, 372, 786–788. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Hughes, S.H. Clonal Expansion of Infected CD4+ T Cells in People Living with HIV. Viruses 2021, 13, 2078. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Gu, D.S.; Kagnoff, M.F. Activating protein-1 cooperates with phorbol ester activation signals to increase HIV-1 expression. Aids 1996, 10, 819–826. [Google Scholar] [CrossRef] [PubMed]
- D’orso, I.; Frankel, A.D. HIV-1 Tat: Its Dependence on Host Factors is Crystal Clear. Viruses 2010, 2, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Karn, J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 454–455, 328–339. [Google Scholar] [CrossRef]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.-M.W.; Margolis, D.M. Chromatin Regulation and the Histone Code in HIV Latency. Yale J. Biol. Med. 2017, 90, 229–243. [Google Scholar]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.-C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-Dependent Regulation of Integrated HIV-1 Expression by Host Gene Transcriptional Readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Paras, P.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Pazin, M.J.; Sheridan, P.L.; Cannon, K.; Cao, Z.; Keck, J.G.; Kadonaga, J.T.; A Jones, K. NF-kappa B-mediated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes Dev. 1996, 10, 37–49. [Google Scholar] [CrossRef]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic Regulation of HIV-1 Latency by Cytosine Methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG Methylation Controls Reactivation of HIV from Latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.-M.; Volker, J.L.; Galvin, K.M.; Davie, J.; Shi, Y.; Hansen, U.; Margolis, D.M. The Human Factors YY1 and LSF Repress the Human Immunodeficiency Virus Type 1 Long Terminal Repeat via Recruitment of Histone Deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- Jiang, G.; Nguyen, D.; Archin, N.M.; Yukl, S.A.; Méndez-Lagares, G.; Tang, Y.; Elsheikh, M.M.; Thompson, G.R.; Hartigan-O’connor, D.J.; Margolis, D.M.; et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J. Clin. Investig. 2018, 128, 1190–1198. [Google Scholar] [CrossRef]
- Peterson, J.; Lewis, C.; Burgos, S.; Manickam, A.; Xu, Y.; Clutton, G.; Simon, J.; Margolis, D.; Goonetilleke, N.; Browne, E.; et al. A histone deacetylase network regulates epigenetic reprogramming and viral silencing in HIV infected cells. bioRxiv 2022, 2022.05. 09.491199. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4+T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef]
- Moron-Lopez, S.; Telwatte, S.; Sarabia, I.; Battivelli, E.; Montano, M.; Macedo, A.B.; Aran, D.; Butte, A.J.; Jones, R.B.; Bosque, A.; et al. Human splice factors contribute to latent HIV infection in primary cell models and blood CD4+ T cells from ART-treated individuals. PLoS Pathog. 2020, 16, e1009060. [Google Scholar] [CrossRef]
- Dufour, C.; Gantner, P.; Fromentin, R.; Chomont, N. The multifaceted nature of HIV latency. J. Clin. Investig. 2020, 130, 3381–3390. [Google Scholar] [CrossRef] [PubMed]
- Verdikt, R.; Hernalsteens, O.; Van Lint, C. Epigenetic Mechanisms of HIV-1 Persistence. Vaccines 2021, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Engel, D.; Mizell, S.B.; Hallahan, C.W.; Fischette, M.; Park, S.; Davey, R.T.; Dybul, M.; Kovacs, J.A.; Metcalf, J.A.; et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 1999, 5, 651–655. [Google Scholar] [CrossRef]
- Dybul, M.; Hidalgo, B.; Chun, T.; Belson, M.; Migueles, S.A.; Justement, J.S.; Herpin, B.; Perry, C.; Hallahan, C.W.; Davey, R.T.; et al. Pilot Study of the Effects of Intermittent Interleukin-2 on Human Immunodeficiency Virus (HIV)–Specific Immune Responses in Patients Treated during Recently Acquired HIV Infection. J. Infect. Dis. 2002, 185, 61–68. [Google Scholar] [CrossRef]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; Berge, I.J.T.; Yong, S.-L.; Fox, C.H.; Roos, M.T.; et al. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. Aids 1999, 13, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Ylisastigui, L.; Coull, J.J.; Rucker, V.C.; Melander, C.; Bosch, R.J.; Brodie, S.J.; Corey, L.; Sodora, D.L.; Dervan, P.B.; Margolis, D.M. Polyamides Reveal a Role for Repression in Latency within Resting T Cells of HIV-Infected Donors. J. Infect. Dis. 2004, 190, 1429–1437. [Google Scholar] [CrossRef][Green Version]
- Ylisastigui, L.; Archin, N.M.; Lehrman, G.; Bosch, R.J.; Margolis, D.M. Coaxing HIV-1 from resting CD4 T cells. Aids 2004, 18, 1101–1108. [Google Scholar] [CrossRef]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; A Spina, C.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef]
- Archin, N.M.; Eron, J.J.; Palmer, S.; Hartmann-Duff, A.; A Martinson, J.; Wiegand, A.; Bandarenko, N.; Schmitz, J.L.; Bosch, R.J.; Landay, A.L.; et al. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. Aids 2008, 22, 1131–1135. [Google Scholar] [CrossRef]
- Sagot-Lerolle, N.; Lamine, A.; Chaix, M.-L.; Boufassa, F.; Aboulker, J.-P.; Costagliola, D.; Goujard, C.; Paller, C.; Delfraissy, J.-F.; Lambotte, O. Prolonged valproic acid treatment does not reduce the size of latent HIV reservoir. Aids 2008, 22, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Lai, J.; Callender, M.; Pitt, E.; Zhang, H.; Margolick, J.B.; Gallant, J.E.; Cofrancesco, J.J.; Moore, R.D.; Gange, S.; et al. Stability of the Latent Reservoir for HIV-1 in Patients Receiving Valproic Acid. J. Infect. Dis. 2007, 195, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Ellenberg, P.; Churchill, M.; Lewin, S.R. HDAC inhibitors in HIV. Immunol. Cell Biol. 2012, 90, 47–54. [Google Scholar] [CrossRef]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef]
- A Rasmussen, T.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Søgaard, O.S.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Østergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development. Hum. Vaccines Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Rasmussen, T.A.; Dinarello, A.C. Histone Deacetylase Inhibitors for Purging HIV-1 from the Latent Reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef]
- Verdikt, R.; Bendoumou, M.; Bouchat, S.; Nestola, L.; Pasternak, A.O.; Darcis, G.; Avettand-Fenoel, V.; Vanhulle, C.; Aït-Ammar, A.; Santangelo, M.; et al. Novel role of UHRF1 in the epigenetic repression of the latent HIV-1. Ebiomedicine 2022, 79, 103985. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.-S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. Aids 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Dashti, A.; Mavigner, M.; Chahroudi, A. Latency Reversal 2.0: Giving the Immune System a Seat at the Table. Curr. HIV/AIDS Rep. 2021, 18, 117–127. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.-F.; Kwon, H.; Fenard, D.; Bisgrove, D.; Verdin, E.; Greene, W.C. Prostratin Antagonizes HIV Latency by Activating NF-κB. J. Biol. Chem. 2004, 279, 42008–42017. [Google Scholar] [CrossRef]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef]
- Brogdon, J.; Ziani, W.; Wang, X.; Veazey, R.S.; Xu, H. In vitro effects of the small-molecule protein kinase C agonists on HIV latency reactivation. Sci. Rep. 2016, 6, 39032. [Google Scholar] [CrossRef]
- Gutiérrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Pérez-Elías, M.J.; Martín, M.E.; Barbas, C.; Ruipérez, J.; Muñoz, E.; Muñoz-Fernández, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Elliott, J.H.; McMahon, J.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Spivak, A.M.; Andrade, A.; Eisele, E.; Hoh, R.; Bacchetti, P.; Bumpus, N.N.; Emad, F.; Buckheit, R., III; McCance-Katz, E.F.; Lai, J.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin. Infect. Dis. 2014, 58, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Dutra, M.S.; Spivak, A.; Marlett, J.M.; Murry, J.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological Activation of Non-canonical NF-κB Signaling Activates Latent HIV-1 Reservoirs In Vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef]
- Lim, S.-Y.; Osuna, C.E.; Hraber, P.T.; Hesselgesser, J.; Gerold, J.M.; Barnes, T.L.; Sanisetty, S.; Seaman, M.S.; Lewis, M.G.; Geleziunas, R.; et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 2018, 10, eaao4521. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.Q.; Alvord, W.G.; Li, Y.; Deleage, C.; Nag, M.; Oswald, K.; Thomas, J.A.; Pyle, C.; Bosche, W.J.; Coalter, V.; et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight 2019, 4, e127717. [Google Scholar] [CrossRef]
- Bekerman, E.; Hesselgesser, J.; Carr, B.; Nagel, M.; Hung, M.; Wang, A.; Stapleton, L.; von Gegerfelt, A.; Elyard, H.A.; Lifson, J.D.; et al. PD-1 Blockade and TLR7 Activation Lack Therapeutic Benefit in Chronic Simian Immunodeficiency Virus-Infected Macaques on Antiretroviral Therapy. Antimicrob. Agents Chemother. 2019, 63, e01163-19. [Google Scholar] [CrossRef]
- Borducchi, E.N.; Liu, J.; Nkolola, J.P.; Cadena, A.M.; Yu, W.-H.; Fischinger, S.; Broge, T.; Abbink, P.; Mercado, N.B.; Chandrashekar, A.; et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 2018, 563, 360–364. [Google Scholar] [CrossRef]
- Vibholm, L.K.; Konrad, C.V.; Schleimann, M.H.; Frattari, G.; Winckelmann, A.; Klastrup, V.; Jensen, N.M.; Jensen, S.S.; Schmidt, M.; Wittig, B.; et al. Effects of 24-week Toll-like receptor 9 agonist treatment in HIV type 1+ individuals. Aids 2019, 33, 1315–1325. [Google Scholar] [CrossRef]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, P.L.H.W.L.S.A.Y.D.V.H.W.C.G.J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kanuma, T.; Takahama, S.; Okamura, T.; Moriishi, E.; Ishii, K.J.; Terahara, K.; Yasutomi, Y. STING agonists activate latently infected cells and enhance SIV-specific responses ex vivo in naturally SIV controlled cynomolgus macaques. Sci. Rep. 2019, 9, 5917. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef]
- Wightman, F.; Solomon, A.; Kumar, S.S.; Urriola, N.; Gallagher, K.; Hiener, B.; Palmer, S.; Mcneil, C.; Garsia, R.; Lewin, S.R. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. Aids 2015, 29, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Gordon, S.; Chan, C.N.; Wang, H.; Lindemuth, E.; Galardi, C.; Falcinelli, S.D.; Raines, S.L.M.; Read, J.L.; Nguyen, K.; et al. CTLA-4 and PD-1 dual blockade induces SIV reactivation without control of rebound after antiretroviral therapy interruption. Nat. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.L.; Bosch, R.J.; McKhann, A.; Moseley, K.F.; Wimbish, C.L.; Hendrickx, S.M.; Messer, M.; Furlong, M.; Campbell, D.M.; Jennings, C.; et al. Suspected Immune-Related Adverse Events With an Anti-PD-1 Inhibitor in Otherwise Healthy People With HIV. J. Acquir. Immune Defic. Syndr. 2021, 87, e234–e236. [Google Scholar] [CrossRef] [PubMed]
- Szaniawski, M.; Spivak, A.M. Senotherapeutics and HIV-1 Persistence. Curr. HIV/AIDS Rep. 2020, 17, 219–225. [Google Scholar] [CrossRef]
- Spivak, A.M.; Larragoite, E.T.; Coletti, M.L.; Macedo, A.B.; Martins, L.J.; Bosque, A.; Planelles, V. Janus kinase inhibition suppresses PKC-induced cytokine release without affecting HIV-1 latency reversal ex vivo. Retrovirology 2016, 13, 88. [Google Scholar] [CrossRef]
- Scripture-Adams, D.D.; Brooks, D.G.; Korin, Y.D.; Zack, J.A. Interleukin-7 Induces Expression of Latent Human Immunodeficiency Virus Type 1 with Minimal Effects on T-Cell Phenotype. J. Virol. 2002, 76, 13077–13082. [Google Scholar] [CrossRef]
- Katlama, C.; Lambert-Niclot, S.; Assoumou, L.; Papagno, L.; Lecardonnel, F.; Zoorob, R.; Tambussi, G.; Clotet, B.; Youle, M.; Achenbach, C.J.; et al. Treatment intensification followed by interleukin-7 reactivates HIV without reducing total HIV DNA. Aids 2016, 30, 221–230. [Google Scholar] [CrossRef]
- Miller, J.S.; Davis, Z.B.; Helgeson, E.; Reilly, C.; Thorkelson, A.; Anderson, J.; Lima, N.S.; Jorstad, S.; Hart, G.T.; Lee, J.H.; et al. Safety and virologic impact of the IL-15 superagonist N-803 in people living with HIV: A phase 1 trial. Nat. Med. 2022, 28, 392–400. [Google Scholar] [CrossRef]
- Kroon, E.D.; Ananworanich, J.; Pagliuzza, A.; Rhodes, A.; Phanuphak, N.; Trautmann, L.; Mitchell, J.L.; Chintanaphol, M.; Intasan, J.; Pinyakorn, S.; et al. A randomized trial of vorinostat with treatment interruption after initiating antiretroviral therapy during acute HIV-1 infection. J. Virus Erad. 2020, 6, 100004. [Google Scholar] [CrossRef] [PubMed]
- Goonetilleke, N.; Clutton, G.; Swanstrom, R.; Joseph, S.B. Blocking Formation of the Stable HIV Reservoir: A New Perspective for HIV-1 Cure. Front. Immunol. 2019, 10, 1966. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01966 (accessed on 4 April 2023). [CrossRef] [PubMed]
- Ellis, J.; Van Maurik, A.; Fortunato, L.; Gisbert, S.; Chen, K.; Schwartz, A.; McHugh, S.; Want, A.; Franco, S.S.; Oliveira, J.-J.; et al. Anti-IL-7 receptor α monoclonal antibody (GSK2618960) in healthy subjects—A randomized, double-blind, placebo-controlled study. Br. J. Clin. Pharmacol. 2019, 85, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Exposito, J.G.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badía, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef]
- Pardons, M.; Fromentin, R.; Pagliuzza, A.; Routy, J.-P.; Chomont, N. Latency-Reversing Agents Induce Differential Responses in Distinct Memory CD4 T Cell Subsets in Individuals on Antiretroviral Therapy. Cell Rep. 2019, 29, 2783–2795.e5. [Google Scholar] [CrossRef]
- Rodari, A.; Darcis, G.; Van Lint, C.M. The Current Status of Latency Reversing Agents for HIV-1 Remission. Annu. Rev. Virol. 2021, 8, 491–514. [Google Scholar] [CrossRef]
- Leth, S.; Schleimann, M.H.; Nissen, S.K.; Højen, J.F.; Olesen, R.; E Graversen, M.; Jørgensen, S.; Kjær, A.S.; Denton, P.W.; Mørk, A.; et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): A single-arm, phase 1B/2A trial. Lancet HIV 2016, 3, e463–e472. [Google Scholar] [CrossRef]
- Gay, C.L.; Kuruc, J.D.; Falcinelli, S.D.; Warren, J.A.; Reifeis, S.A.; Kirchherr, J.L.; James, K.S.; Dewey, M.G.; Helms, A.; Allard, B.; et al. Assessing the impact of AGS-004, a dendritic cell-based immunotherapy, and vorinostat on persistent HIV-1 Infection. Sci. Rep. 2020, 10, 5134. [Google Scholar] [CrossRef]
- Fidler, S.; Stöhr, W.; Pace, M.; Dorrell, L.; Lever, A.; Pett, S.; Loes, S.K.-D.; Fox, J.; Clarke, A.; Nelson, M.; et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): A phase 2, randomised trial. Lancet 2020, 395, 888–898. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, C.A.; Margolis, D.M.; Browne, E.P. New Concepts in Therapeutic Manipulation of HIV-1 Transcription and Latency: Latency Reversal versus Latency Prevention. Viruses 2023, 15, 1677. https://doi.org/10.3390/v15081677
Lewis CA, Margolis DM, Browne EP. New Concepts in Therapeutic Manipulation of HIV-1 Transcription and Latency: Latency Reversal versus Latency Prevention. Viruses. 2023; 15(8):1677. https://doi.org/10.3390/v15081677
Chicago/Turabian StyleLewis, Catherine A., David M. Margolis, and Edward P. Browne. 2023. "New Concepts in Therapeutic Manipulation of HIV-1 Transcription and Latency: Latency Reversal versus Latency Prevention" Viruses 15, no. 8: 1677. https://doi.org/10.3390/v15081677
APA StyleLewis, C. A., Margolis, D. M., & Browne, E. P. (2023). New Concepts in Therapeutic Manipulation of HIV-1 Transcription and Latency: Latency Reversal versus Latency Prevention. Viruses, 15(8), 1677. https://doi.org/10.3390/v15081677