
A CRISPR Screen of HIV Dependency Factors Reveals That CCNT1 Is Non-Essential in T Cells but Required for HIV-1 Reactivation from Latency

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Culture and Maintenance
2.2. HIV-CRISPR Library Transduction and Virus-Encapsidated CRISPR Guide Screening
2.3. Latency HIV-CRISPR Screen
2.4. Validation of Screen Hits
2.5. Protein Isolation and Western Blotting
2.6. Genomic Editing Analysis
2.7. LRA Treatments
2.8. Primary CD4+ Cell Isolation and Latency Model
2.9. Primary CD4+ T Cell Activation Test
2.10. RNA-seq Analysis of CCNT1 Knockout Cells
2.11. RNAseq Analysis Methods
3. Results
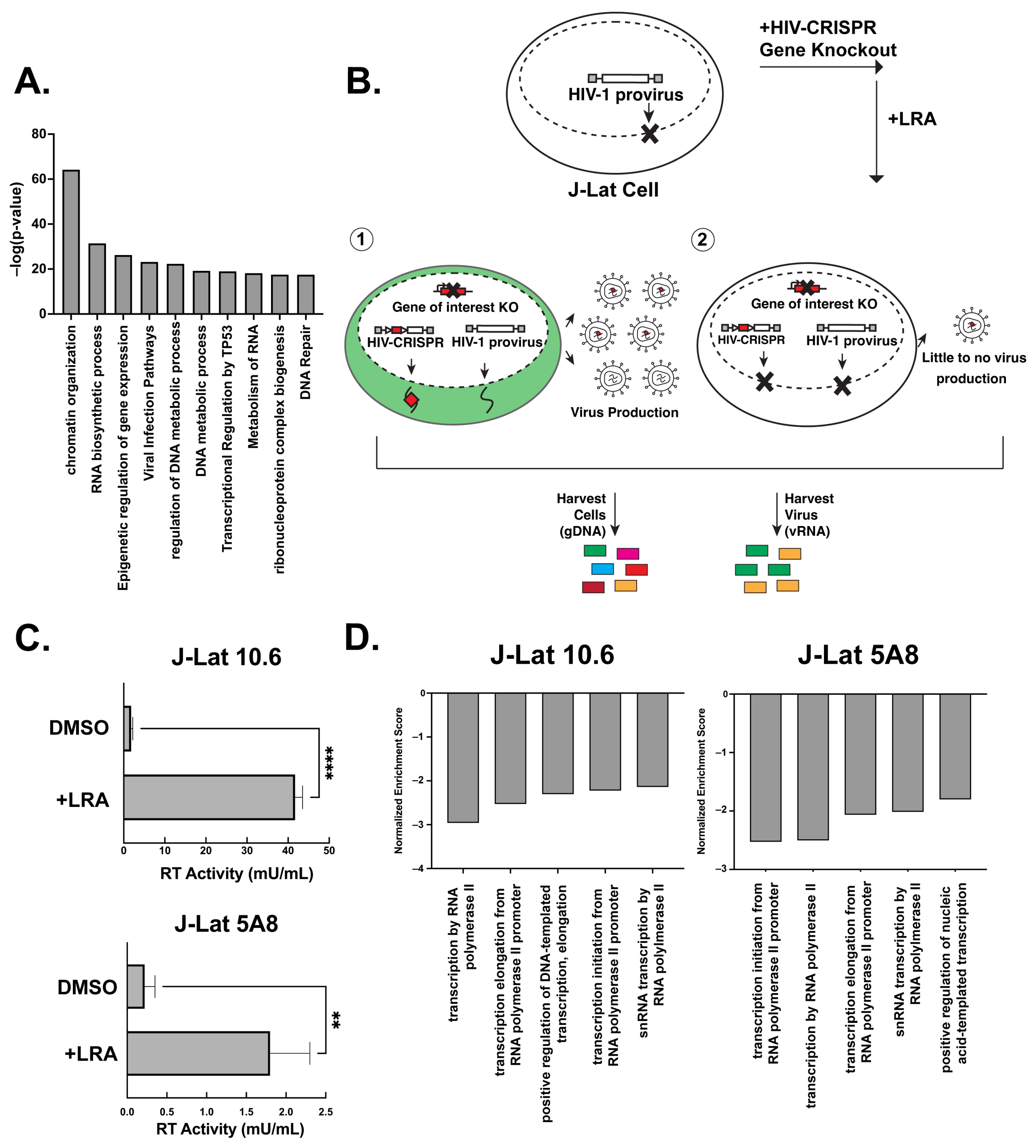
3.1. A Latency HIV-CRISPR Screen of HIV Dependency Factors to Identify Latency Reversal Factors
3.2. Cyclin T1 Is Essential for Reactivation from Latency in Both J-Lat and Primary T Cells
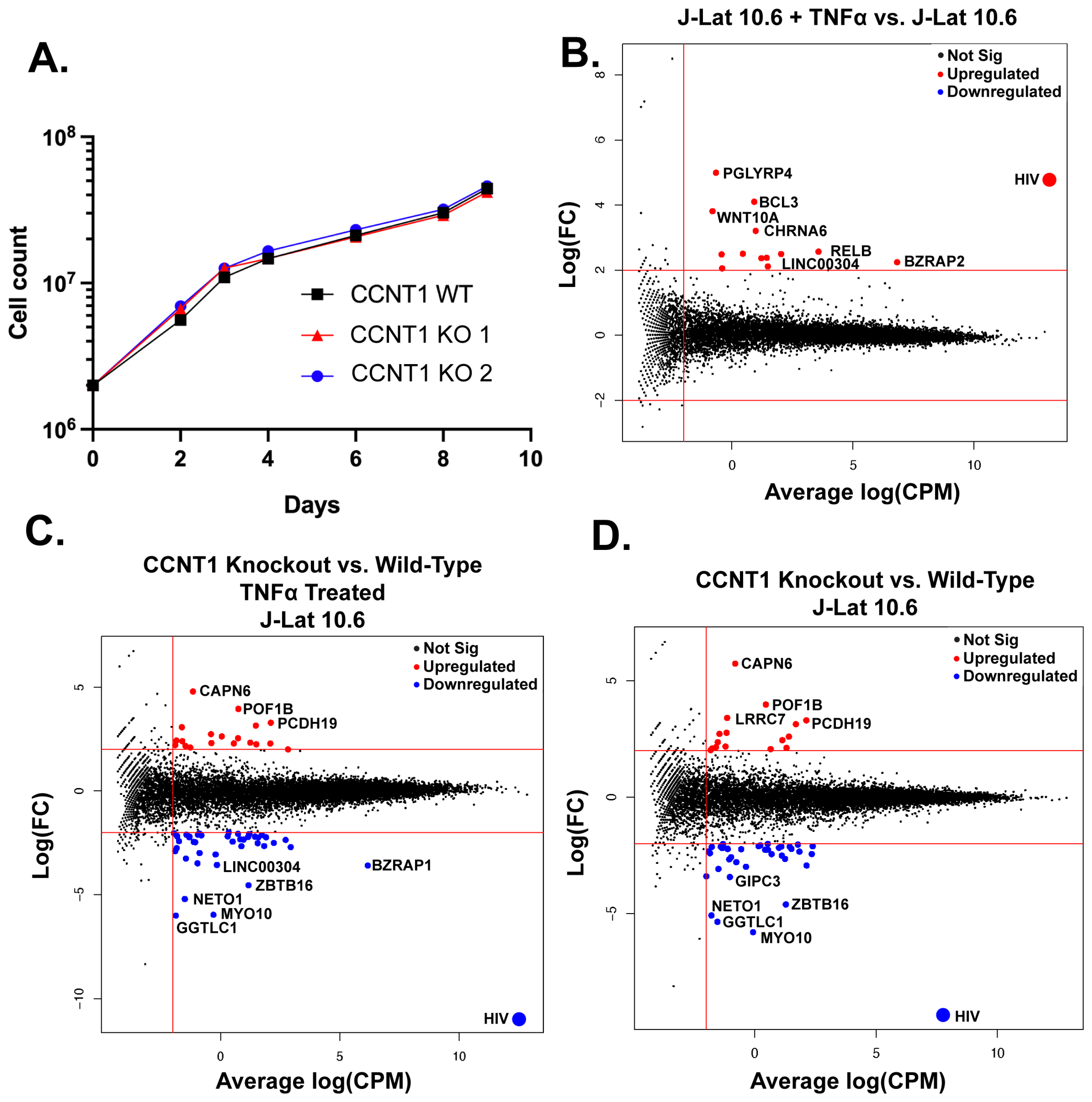
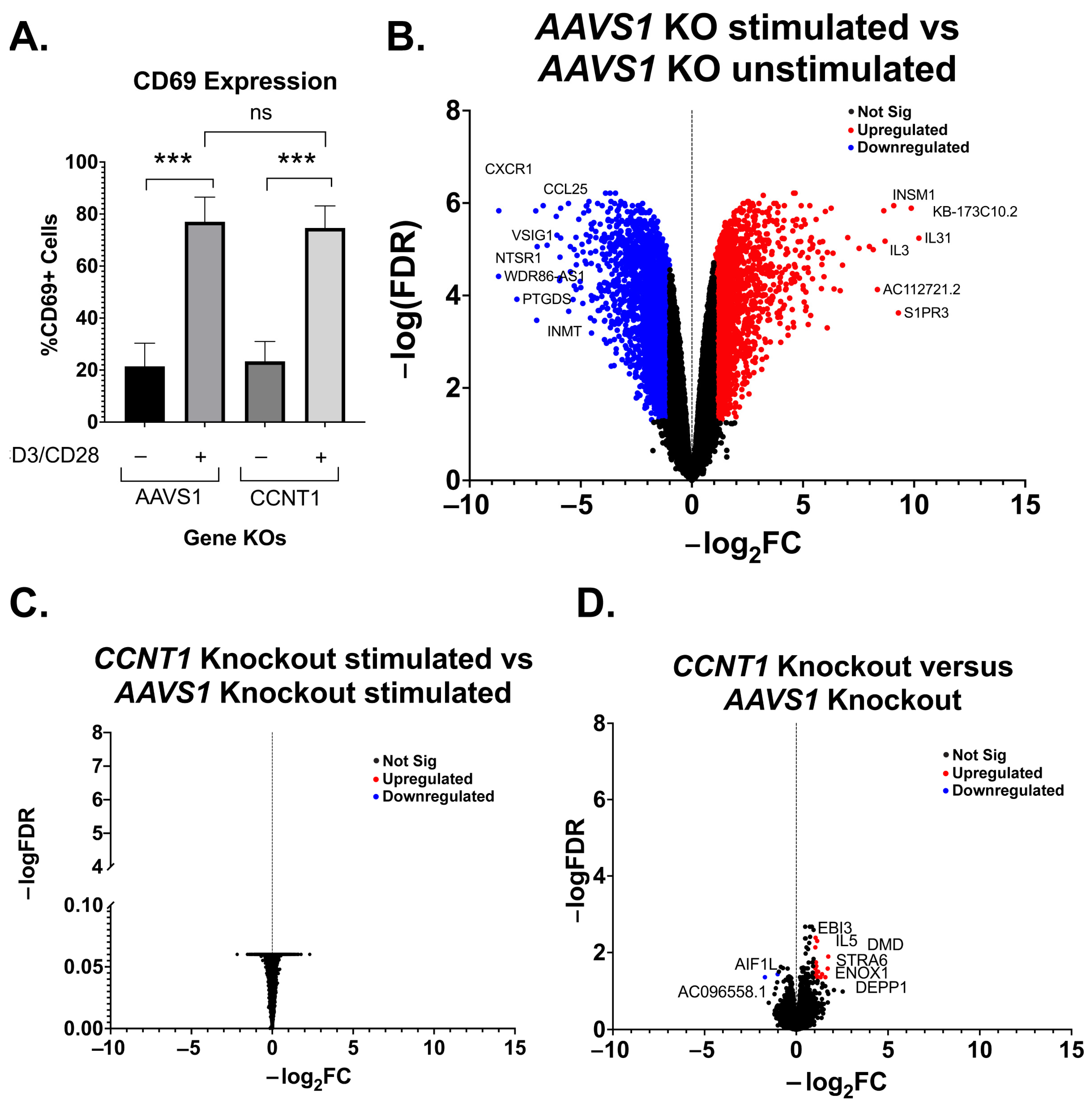
3.3. Cyclin T1 Is Non-Essential in T Cells and Regulates Host Genes to a Much Lesser Extent than It Regulates HIV-1
4. Discussion
4.1. Cyclin T1 Is Much More Important for HIV Latency Reversal than T Cell Biology In Vitro
4.2. Other Hits in the HIV-CRISPR Screen
4.3. HIV Dependency Factors Versus Host Genes Necessary for Latency Reversal
4.4. Gene Paralogs in a “Block and Lock” Latency Approach
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Rodari, A.; Darcis, G.; Van Lint, C.M. The Current Status of Latency Reversing Agents for HIV-1 Remission. Annu. Rev. Virol. 2021, 8, 491–514. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Grau-Exposito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badia, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef]
- Li, C.; Mori, L.; Valente, S.T. The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin A. J. Infect. Dis. 2021, 223, 46–53. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio 2019, 10, e02662-18. [Google Scholar] [CrossRef]
- Turner, A.M.; Ackley, A.M.; Matrone, M.A.; Morris, K.V. Characterization of an HIV-targeted transcriptional gene-silencing RNA in primary cells. Hum. Gene Ther. 2012, 23, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol. Ther. Nucleic Acids 2015, 4, e261. [Google Scholar] [CrossRef]
- Marconi, V.C.; Moser, C.; Gavegnano, C.; Deeks, S.G.; Lederman, M.M.; Overton, E.T.; Tsibris, A.; Hunt, P.W.; Kantor, A.; Sekaly, R.P.; et al. Randomized Trial of Ruxolitinib in Antiretroviral-Treated Adults with Human Immunodeficiency Virus. Clin. Infect. Dis. 2022, 74, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.; Janssens, D.H.; Paddison, P.J.; Browne, E.P.; Henikoff, S.; OhAinle, M.; Emerman, M. A modular CRISPR screen identifies individual and combination pathways contributing to HIV-1 latency. PLoS Pathog. 2023, 19, e1011101. [Google Scholar] [CrossRef]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2019, 10, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Taube, R.; Fujinaga, K.; Peterlin, B.M. P-TEFb containing cyclin K and Cdk9 can activate transcription via RNA. J. Biol. Chem. 2002, 277, 16873–16878. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Analysis of the effect of natural sequence variation in Tat and in cyclin T on the formation and RNA binding properties of Tat-cyclin T complexes. J. Virol. 1999, 73, 5777–5786. [Google Scholar] [CrossRef]
- Montoya, V.R.; Ready, T.M.; Felton, A.; Fine, S.R.; OhAinle, M.; Emerman, M. A Virus-Packageable CRISPR System Identifies Host Dependency Factors Co-Opted by Multiple HIV-1 Strains. mBio 2023, 14, e0000923. [Google Scholar] [CrossRef]
- Roesch, F.; OhAinle, M. HIV-CRISPR: A CRISPR/Cas9 Screening Method to Identify Genes Affecting HIV Replication. Bio-Protocol 2020, 10, e3614. [Google Scholar] [CrossRef]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.H.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019, 14, 756–780. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sanchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 184, 120–132.e114. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, J.; Naessens, E.; Vanderstraeten, H.; Landi, A.; Iannucci, V.; Van Nuffel, A.; Taghon, T.; Pizzato, M.; Verhasselt, B. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE 2012, 7, e50859. [Google Scholar] [CrossRef] [PubMed]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Humbert, O.; Gisch, D.W.; Wohlfahrt, M.E.; Adams, A.B.; Greenberg, P.D.; Schmitt, T.M.; Trobridge, G.D.; Kiem, H.P. Development of Third-generation Cocal Envelope Producer Cell Lines for Robust Lentiviral Gene Transfer into Hematopoietic Stem Cells and T-cells. Mol. Ther. 2016, 24, 1237–1246. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 2018, 7, e39823. [Google Scholar] [CrossRef]
- Falcinelli, S.D.; Peterson, J.J.; Turner, A.W.; Irlbeck, D.; Read, J.; Raines, S.L.; James, K.S.; Sutton, C.; Sanchez, A.; Emery, A.; et al. Combined noncanonical NF-kappaB agonism and targeted BET bromodomain inhibition reverse HIV latency ex vivo. J. Clin. Investig. 2022, 132, e157281. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef]
- Ivanov, D.; Kwak, Y.T.; Nee, E.; Guo, J.; Garcia-Martinez, L.F.; Gaynor, R.B. Cyclin T1 domains involved in complex formation with Tat and TAR RNA are critical for tat-activation. J. Mol. Biol. 1999, 288, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A. Taking a new TAK on tat transactivation. Genes Dev. 1997, 11, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Peterlin, B.M. Control of RNA initiation and elongation at the HIV-1 promoter. Annu. Rev. Biochem. 1994, 63, 717–743. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Areces, L.B.; Bahador, A.; Mischak, H.; Goodnight, J.; Mushinski, J.F.; Blumberg, P.M. Characterization of ligand and substrate specificity for the calcium-dependent and calcium-independent protein kinase C isozymes. Mol. Pharmacol. 1993, 44, 298–307. [Google Scholar] [PubMed]
- Sung, T.L.; Rice, A.P. Effects of prostratin on Cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 2006, 3, 66. [Google Scholar] [CrossRef]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef]
- Fujinaga, K.; Huang, F.; Peterlin, B.M. P-TEFb: The master regulator of transcription elongation. Mol. Cell 2023, 83, 393–403. [Google Scholar] [CrossRef]
- Bauer, D.; Mazzio, E.; Hilliard, A.; Oriaku, E.T.; Soliman, K.F.A. Effect of apigenin on whole transcriptome profile of TNFalpha-activated MDA-MB-468 triple negative breast cancer cells. Oncol. Lett. 2020, 19, 2123–2132. [Google Scholar]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Palmer, S.; Chen, Y.H. Bcl-3, a multifaceted modulator of NF-kappaB-mediated gene transcription. Immunol. Res. 2008, 42, 210–218. [Google Scholar] [CrossRef]
- Dillon, S.R.; Sprecher, C.; Hammond, A.; Bilsborough, J.; Rosenfeld-Franklin, M.; Presnell, S.R.; Haugen, H.S.; Maurer, M.; Harder, B.; Johnston, J.; et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 2004, 5, 752–760. [Google Scholar] [CrossRef]
- Kohoutek, J.; Li, Q.; Blazek, D.; Luo, Z.; Jiang, H.; Peterlin, B.M. Cyclin T2 is essential for mouse embryogenesis. Mol. Cell. Biol. 2009, 29, 3280–3285. [Google Scholar] [CrossRef]
- Oven, I.; Brdickova, N.; Kohoutek, J.; Vaupotic, T.; Narat, M.; Peterlin, B.M. AIRE recruits P-TEFb for transcriptional elongation of target genes in medullary thymic epithelial cells. Mol. Cell. Biol. 2007, 27, 8815–8823. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Yu, W.; Rice, A.P. Limited redundancy in genes regulated by Cyclin T2 and Cyclin T1. BMC Res. Notes 2011, 4, 260. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Groffen, J.; Warburton, D.; Sneddon, T.P. The human gamma-glutamyltransferase gene family. Hum. Genet. 2008, 123, 321–332. [Google Scholar] [CrossRef]
- Berg, J.S.; Derfler, B.H.; Pennisi, C.M.; Corey, D.P.; Cheney, R.E. Myosin-X, a novel myosin with pleckstrin homology domains, associates with regions of dynamic actin. J. Cell. Sci. 2000, 113, 3439–3451. [Google Scholar] [CrossRef]
- Uhl, J.; Gujarathi, S.; Waheed, A.A.; Gordon, A.; Freed, E.O.; Gousset, K. Myosin-X is essential to the intercellular spread of HIV-1 Nef through tunneling nanotubes. J. Cell Commun. Signal. 2019, 13, 209–224. [Google Scholar] [CrossRef]
- Zhang, S.; Laouar, A.; Denzin, L.K.; Sant’Angelo, D.B. Zbtb16 (PLZF) is stably suppressed and not inducible in non-innate T cells via T cell receptor-mediated signaling. Sci. Rep. 2015, 5, 12113. [Google Scholar] [CrossRef]
- Yasuda-Yamahara, M.; Rogg, M.; Yamahara, K.; Maier, J.I.; Huber, T.B.; Schell, C. AIF1L regulates actomyosin contractility and filopodial extensions in human podocytes. PLoS ONE 2018, 13, e0200487. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef]
- Garriga, J.; Peng, J.; Parreno, M.; Price, D.H.; Henderson, E.E.; Grana, X. Upregulation of cyclin T1/CDK9 complexes during T cell activation. Oncogene 1998, 17, 3093–3102. [Google Scholar] [CrossRef]
- Huang, F.; Nguyen, T.T.; Echeverria, I.; Rakesh, R.; Cary, D.C.; Paculova, H.; Sali, A.; Weiss, A.; Peterlin, B.M.; Fujinaga, K. Reversible phosphorylation of cyclin T1 promotes assembly and stability of P-TEFb. eLife 2021, 10, e68473. [Google Scholar] [CrossRef]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998, 17, 7056–7065. [Google Scholar] [CrossRef]
- Garber, M.E.; Wei, P.; KewalRamani, V.N.; Mayall, T.P.; Herrmann, C.H.; Rice, A.P.; Littman, D.R.; Jones, K.A. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998, 12, 3512–3527. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lane, W.S.; Fischle, W.; Verdin, E.; Lazar, M.A.; Shiekhattar, R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000, 14, 1048–1057. [Google Scholar] [CrossRef]
- Ning, L.; Rui, X.; Bo, W.; Qing, G. The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury. Cell Death Dis. 2021, 12, 734. [Google Scholar] [CrossRef]
- Yoon, H.G.; Chan, D.W.; Huang, Z.Q.; Li, J.; Fondell, J.D.; Qin, J.; Wong, J. Purification and functional characterization of the human N-CoR complex: The roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003, 22, 1336–1346. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Dai, W.; Wu, F.; McMyn, N.; Song, B.; Walker-Sperling, V.E.; Varriale, J.; Zhang, H.; Barouch, D.H.; Siliciano, J.D.; Li, W.; et al. Genome-wide CRISPR screens identify combinations of candidate latency reversing agents for targeting the latent HIV-1 reservoir. Sci. Transl. Med. 2022, 14, eabh3351. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef]
- Liang, W.S.; Maddukuri, A.; Teslovich, T.M.; de la Fuente, C.; Agbottah, E.; Dadgar, S.; Kehn, K.; Hautaniemi, S.; Pumfery, A.; Stephan, D.A.; et al. Therapeutic targets for HIV-1 infection in the host proteome. Retrovirology 2005, 2, 20. [Google Scholar] [CrossRef][Green Version]
- Hyrcza, M.D.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Heisler, L.; Yang, S.; Wilkins, O.; Ostrowski, M.; Der, S.D. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J. Virol. 2007, 81, 3477–3486. [Google Scholar] [CrossRef]
- Dixit, U.; Bhutoria, S.; Wu, X.; Qiu, L.; Spira, M.; Mathew, S.; Harris, R.; Adams, L.J.; Cahill, S.; Pathak, R.; et al. INI1/SMARCB1 Rpt1 domain mimics TAR RNA in binding to integrase to facilitate HIV-1 replication. Nat. Commun. 2021, 12, 2743. [Google Scholar] [CrossRef]
- Parrish, P.C.R.; Thomas, J.D.; Gabel, A.M.; Kamlapurkar, S.; Bradley, R.K.; Berger, A.H. Discovery of synthetic lethal and tumor suppressor paralog pairs in the human genome. Cell Rep. 2021, 36, 109597. [Google Scholar] [CrossRef]
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription. mBio 2019, 10, e01750-18. [Google Scholar] [CrossRef]
- Rice, A.P. Unexpected Mutations in HIV-1 That Confer Resistance to the Tat Inhibitor Didehydro-Cortistatin A. mBio 2019, 10, e01547-19. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafer, T.L.; Felton, A.; Delgado, Y.; Srinivasan, H.; Emerman, M. A CRISPR Screen of HIV Dependency Factors Reveals That CCNT1 Is Non-Essential in T Cells but Required for HIV-1 Reactivation from Latency. Viruses 2023, 15, 1863. https://doi.org/10.3390/v15091863
Hafer TL, Felton A, Delgado Y, Srinivasan H, Emerman M. A CRISPR Screen of HIV Dependency Factors Reveals That CCNT1 Is Non-Essential in T Cells but Required for HIV-1 Reactivation from Latency. Viruses. 2023; 15(9):1863. https://doi.org/10.3390/v15091863
Chicago/Turabian StyleHafer, Terry L., Abby Felton, Yennifer Delgado, Harini Srinivasan, and Michael Emerman. 2023. "A CRISPR Screen of HIV Dependency Factors Reveals That CCNT1 Is Non-Essential in T Cells but Required for HIV-1 Reactivation from Latency" Viruses 15, no. 9: 1863. https://doi.org/10.3390/v15091863
APA StyleHafer, T. L., Felton, A., Delgado, Y., Srinivasan, H., & Emerman, M. (2023). A CRISPR Screen of HIV Dependency Factors Reveals That CCNT1 Is Non-Essential in T Cells but Required for HIV-1 Reactivation from Latency. Viruses, 15(9), 1863. https://doi.org/10.3390/v15091863