
SARS-CoV-2 Recombination and Coinfection Events Identified in Clinical Samples in Russia
 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
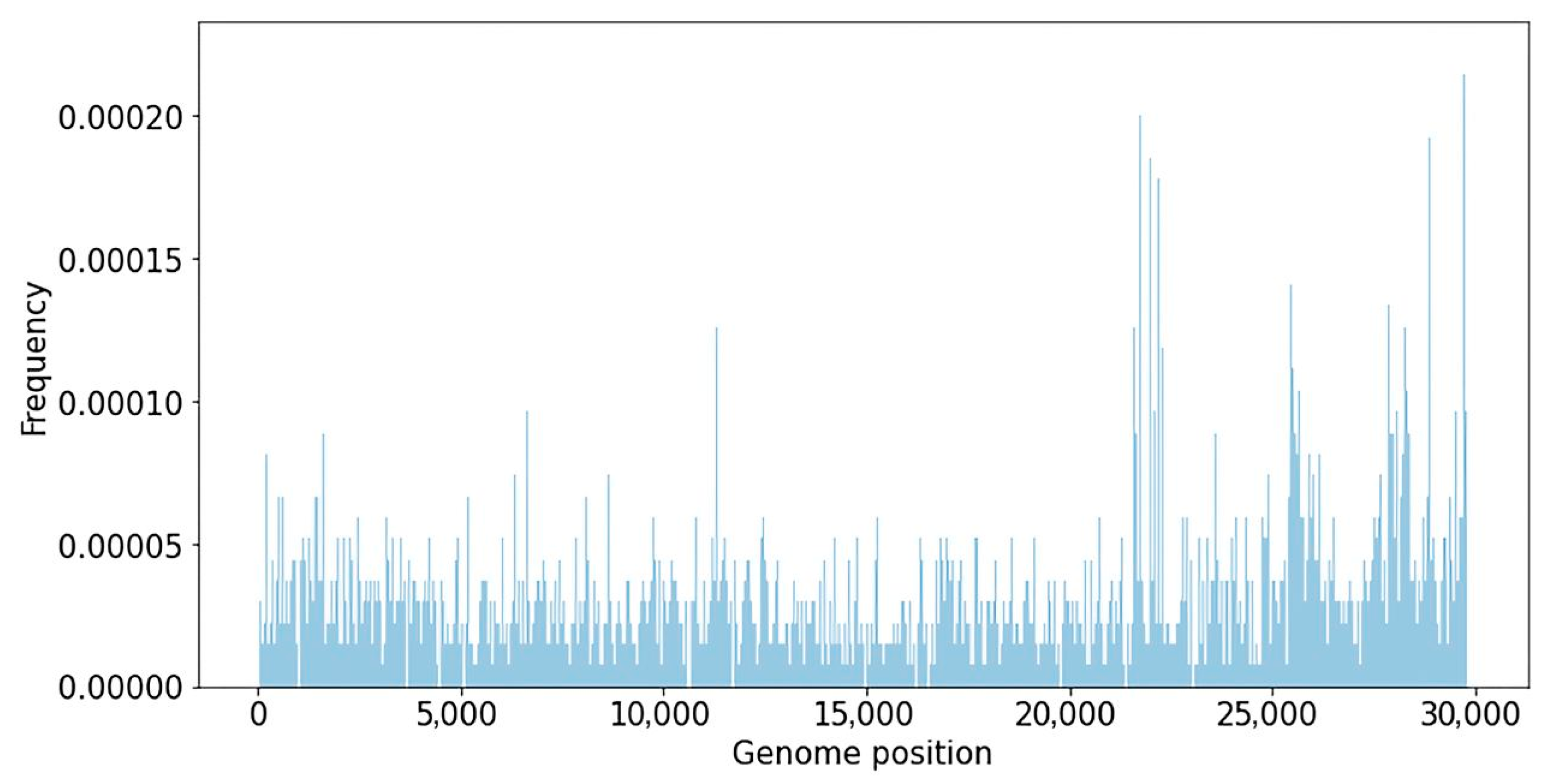
2.1. Database of SARS-CoV-2 Nucleotide Variants
2.2. Samples Collection
2.3. SARS-CoV-2 Whole Genome Sequencing
2.4. SARS-CoV-2 Sequencing of Target Regions
2.5. Consensus Sequences and Genomic Variant Identification
2.6. Data Preparation and Phylogenetic Analysis
2.7. Principal Component Analysis
- Using the VCF files, a table was assembled containing information on the substitutions found in each of the samples;
- Filtering of substitutions in low-coverage regions (<10 reads) of the genome was carried out;
- We removed all substitutions that were not in the reference database, as well as substitutions whose occurrence count was less than 10;
- For each sample, a binary replacement vector was created (each element of the vector corresponds to a certain replacement. The value of the element is 1 if the replacement is present in the sample and 0 if it is absent);
- Using the obtained vectors, the principal component analysis was carried out and implemented in the scikit-learn library of the Python language;
- Visualization of the result was performed using the seaborn library.
2.8. Algorithm for the Detection of Recombinant Genomes and Co-infections
3. Results
3.1. SARS-CoV-2 Nucleotide Variations Database
3.2. SARS-CoV-2 Strain Genomic Classification Using PCA Method
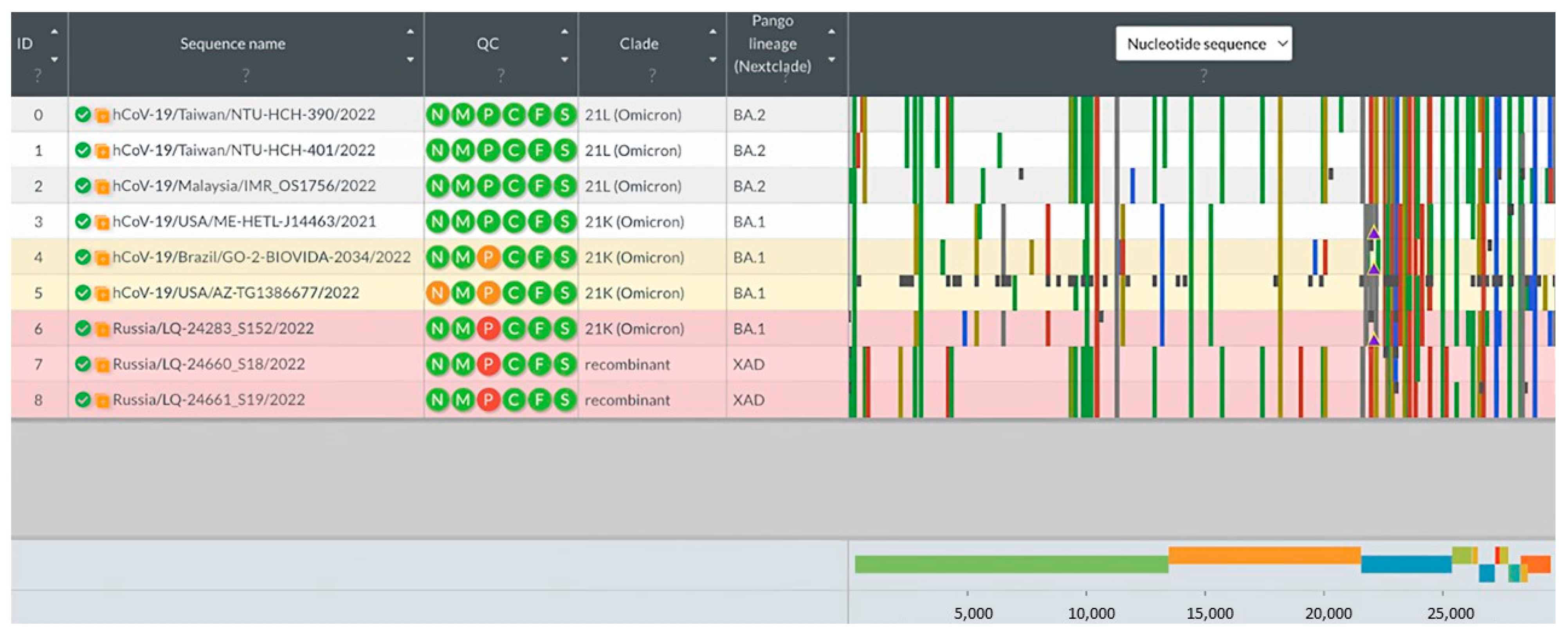
3.3. Analysis of Potential Recombinant Genomes
3.3.1. Analysis of Consensus Genomic Sequences
3.3.2. Analysis of Short Read Alignment Files
3.3.3. Analysis of Nanopore Sequencing Data
3.3.4. Phylogenetic Analysis
3.4. Detection of Co-infections in Sequencing Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Varabyou, A.; Pockrandt, C.; Salzberg, S.L.; Pertea, M. Rapid detection of inter-clade recombination in SARS-CoV-2 with Bolotie. Genetics 2021, 218, iyab074. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e8. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ma, W.; Dang, S.; Chen, L.; Zhang, R.; Mei, S.; Wei, X.; Lv, Q.; Peng, B.; Chen, J.; et al. Possible recombination between two variants of concern in a COVID-19 patient. Emerg. Microbes Infect. 2022, 11, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Lacek, K.A.; Rambo-Martin, B.L.; Batra, D.; Zheng, X.Y.; Sakaguchi, H.; Peacock, T.; Keller, M.; Wilson, M.M.; Sheth, M.; Davis, M.L. Identification of a Novel SARS-CoV-2 Delta-Omicron Recombinant Virus in the United States. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bolze, A.; Basler, T.; White, S.; Dei Rossi, A.; Wyman, D.; Dai, H.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Beatty, M.; et al. Evidence for SARS-CoV-2 Delta and Omicron Co-Infections and Recombination. Infect. Dis. (Except. HIV/AIDS) 2022. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Dhama, K. Recombinant SARS-CoV-2 variants XD, XE, and XF: The emergence of recombinant variants requires an urgent call for research—Correspondence. Int. J. Surg. Lond. Engl. 2022, 102, 106670. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.39972013. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet—Next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- PRINSEQ. Available online: http://prinseq.sourceforge.net (accessed on 15 October 2022).
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Vari-Ant Discovery to Tens of Thousands of Samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- FastTree 2.1.11. Available online: http://www.microbesonline.org/fasttree/ (accessed on 17 September 2022).
- FigTree. Available online: https://github.com/rambaut/figtree/ (accessed on 17 September 2022).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 1 November 2022).
- Nikolaidis, M.; Markoulatos, P.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and Nonhomologous Recombination in Coronavirus Genomes. Mol. Biol. Evol. 2022, 39, msab292. [Google Scholar] [CrossRef] [PubMed]
- TAG-VE Statement on Omicron Sublineages BQ.1 and XBB. Available online: https://www.who.int/news/item/27-10-2022-tag-ve-statement-on-omicron-sublineages-bq.1-and-xbb (accessed on 1 November 2022).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Detected Genetic Lines | Date of Sampling |
|---|---|---|
| LQ-23247 | Delta/21J, Omicron/BA.1 | 29 January 2022 |
| LQ-21013 | Omicron/BA.1, Omicron/BA.2 | 08 January 2022 |
| LQ-23061 | Omicron/BA.1, Omicron/BA.2 | 26 January 2022 |
| LQ-23066 | Omicron/BA.1, Omicron/BA.2 | 26 January 2022 |
| LQ-21871 | Omicron/BA.1, Omicron/BA.2 | 19 January 2022 |
| LQ-22654 | Delta/21J, Omicron/BA.1 | 23 January 2022 |
| LQ-24255 | Omicron/BA.2, Omicron/B.1.1.529 | 22 February 2022 |
| GCG-1605 | Delta/21J, Omicron/BA.1 | 25 February 2022 |
| LQ-3635 | Alpha/B.1.1.7, Beta/B.1.351 | 02 May 2021 |
| LQ-3188 | Alpha/B.1.1.7, Beta/B.1.351 | 23 April 2021 |
| LQ-3937 | B.1.1, Delta/AY.122.4 | 07 May 2021 |
| LQ-6578 | Delta/AY.17, Delta/AY.121.1 | 01 July 2021 |
| LQ-6061 | Alpha/B.1.1.7, Delta/B.1.617.2 | 28 June 2021 |
| KAB | B.1.232, B.1.1.523 | 05 April 2021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernyaeva, E.N.; Ayginin, A.A.; Kosenkov, A.V.; Romanova, S.V.; Tsypkina, A.V.; Luparev, A.R.; Stetsenko, I.F.; Gnusareva, N.I.; Matsvay, A.D.; Savochkina, Y.A.; et al. SARS-CoV-2 Recombination and Coinfection Events Identified in Clinical Samples in Russia. Viruses 2023, 15, 1660. https://doi.org/10.3390/v15081660
Chernyaeva EN, Ayginin AA, Kosenkov AV, Romanova SV, Tsypkina AV, Luparev AR, Stetsenko IF, Gnusareva NI, Matsvay AD, Savochkina YA, et al. SARS-CoV-2 Recombination and Coinfection Events Identified in Clinical Samples in Russia. Viruses. 2023; 15(8):1660. https://doi.org/10.3390/v15081660
Chicago/Turabian StyleChernyaeva, Ekaterina N., Andrey A. Ayginin, Alexey V. Kosenkov, Svetlana V. Romanova, Anastasia V. Tsypkina, Andrey R. Luparev, Ivan F. Stetsenko, Natalia I. Gnusareva, Alina D. Matsvay, Yulia A. Savochkina, and et al. 2023. "SARS-CoV-2 Recombination and Coinfection Events Identified in Clinical Samples in Russia" Viruses 15, no. 8: 1660. https://doi.org/10.3390/v15081660
APA StyleChernyaeva, E. N., Ayginin, A. A., Kosenkov, A. V., Romanova, S. V., Tsypkina, A. V., Luparev, A. R., Stetsenko, I. F., Gnusareva, N. I., Matsvay, A. D., Savochkina, Y. A., & Shipulin, G. A. (2023). SARS-CoV-2 Recombination and Coinfection Events Identified in Clinical Samples in Russia. Viruses, 15(8), 1660. https://doi.org/10.3390/v15081660