
Equine Polyclonal Antibodies Prevent Acute Chikungunya Virus Infection in Mice

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Ethics Statement
2.2. Equine Immunization, Plasmapheresis, Hyperimmune Product Manufacturing
2.2.1. Immunogen
2.2.2. Hyperimmunization
2.2.3. CHIKV-EIG Manufacturing
2.3. Assays
2.3.1. Plaque Assays
2.3.2. Plaque Reduction Neutralization Tests
2.3.3. Infectious Cell Culture Assays
2.3.4. Cytokine Assays
2.4. Animals
2.5. Therapeutic Evaluation of CHIKV-EIG in Immunocompromised Mice
Study Design
2.6. Therapeutic Evaluation of CHIKV-EIG in Immunocompetent Mice
Study Design
2.7. Statistical Analyses
3. Results
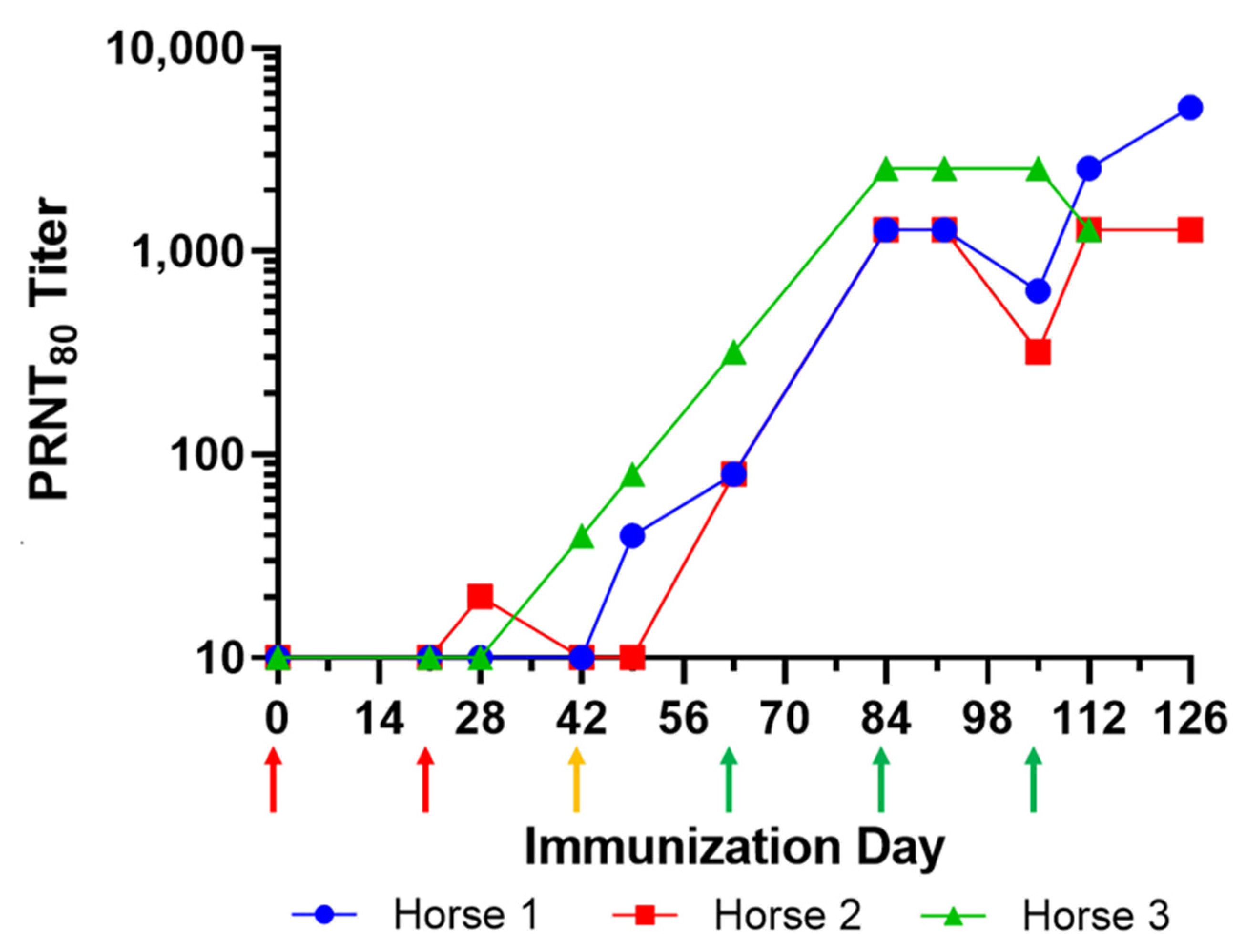
3.1. Immunization of Horses and Manufacturing of Equine CHIKV-EIG Product
3.2. In Vitro Characterization of CHIKV-EIG for Broad-Spectrum Activity
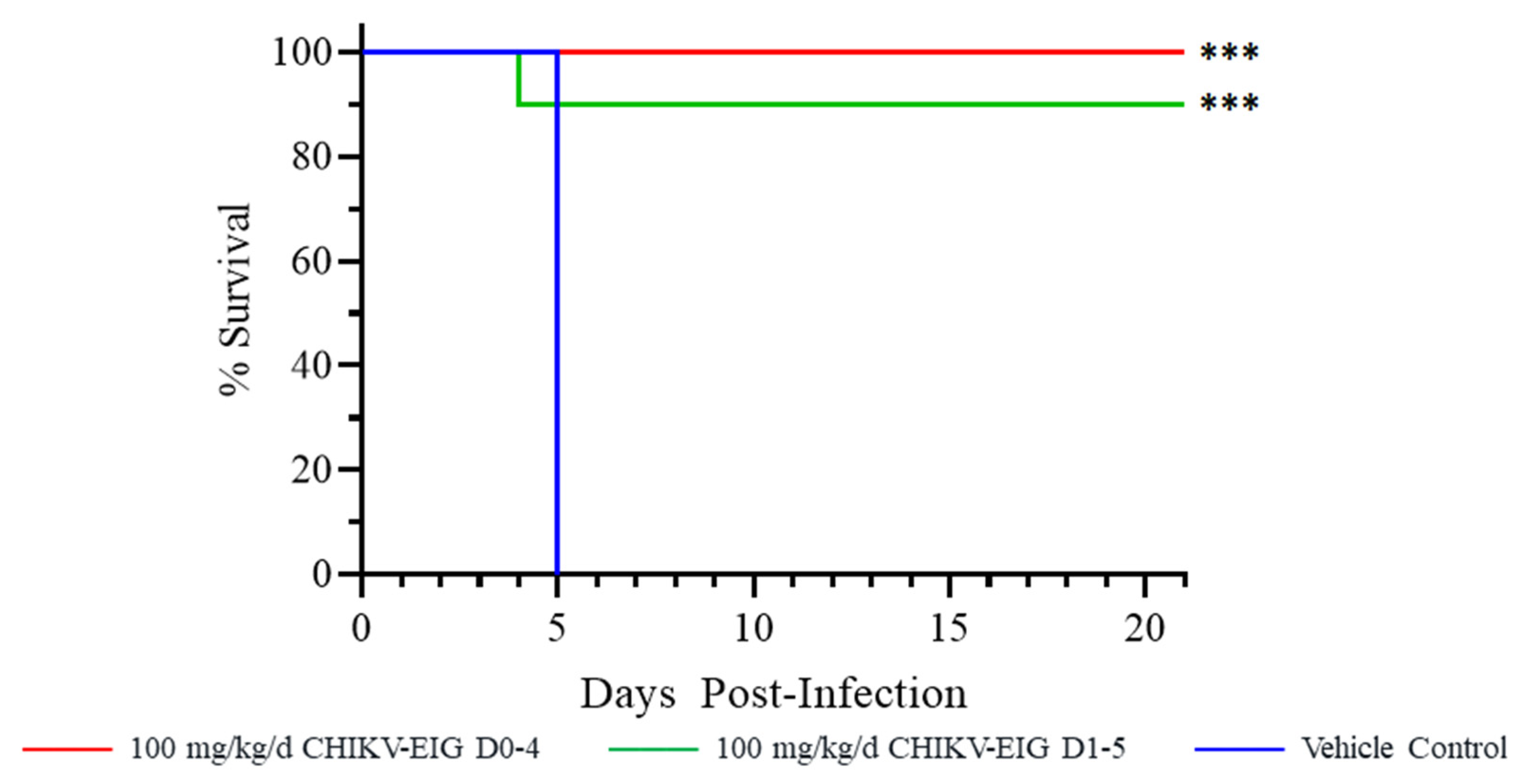
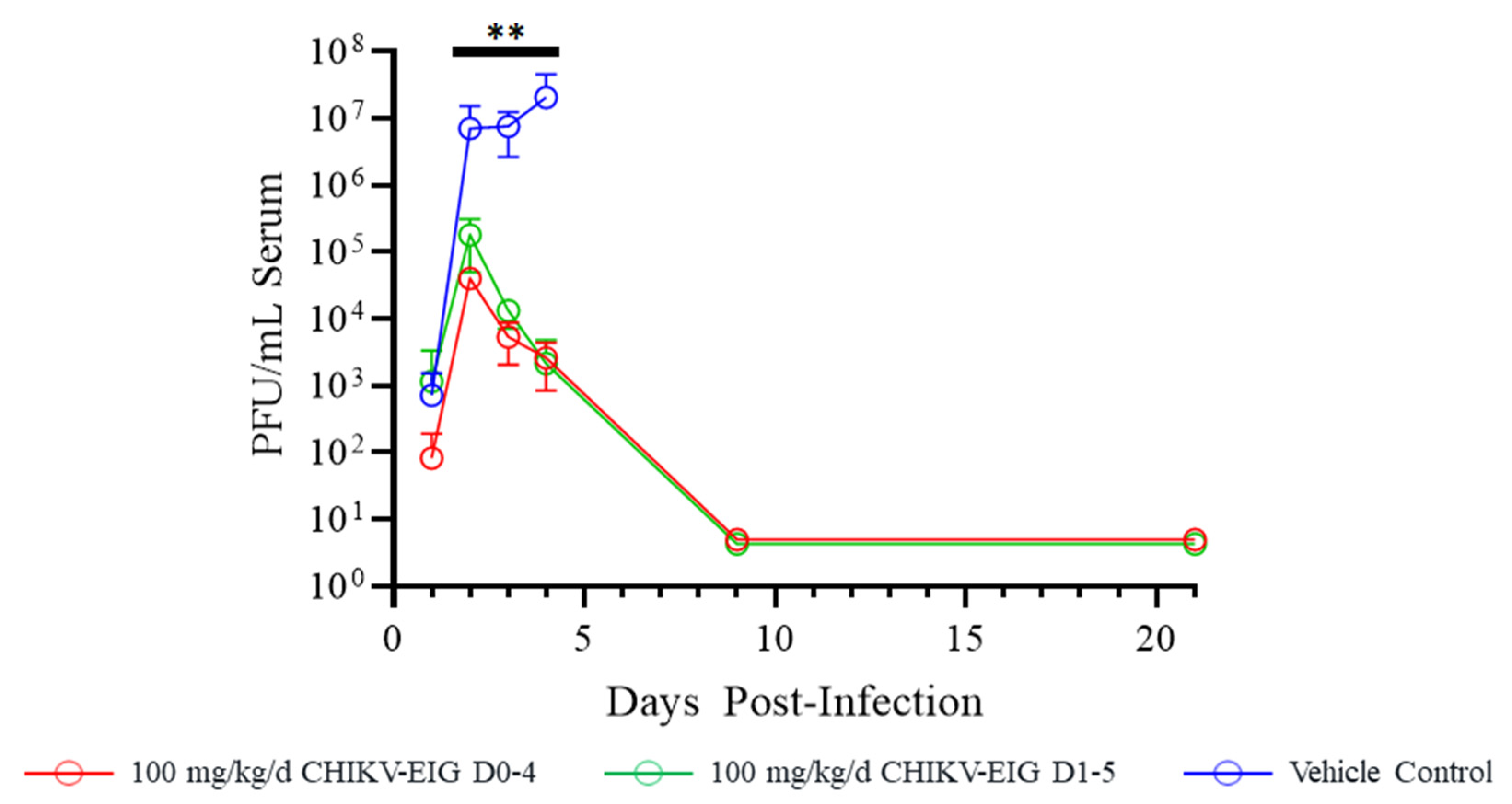
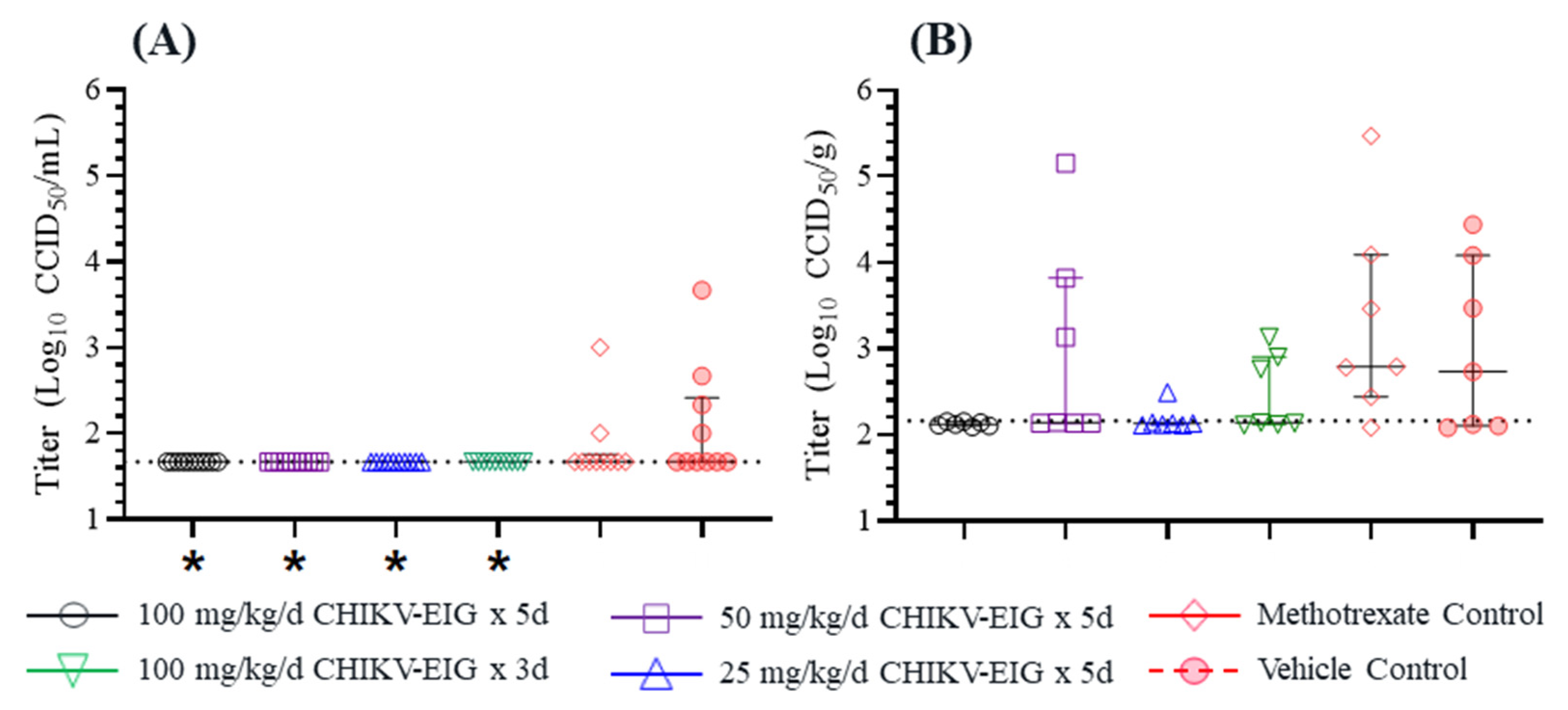
3.3. Protective Efficacy of CHIKV-EIG in an Immunocompromised Mouse Model
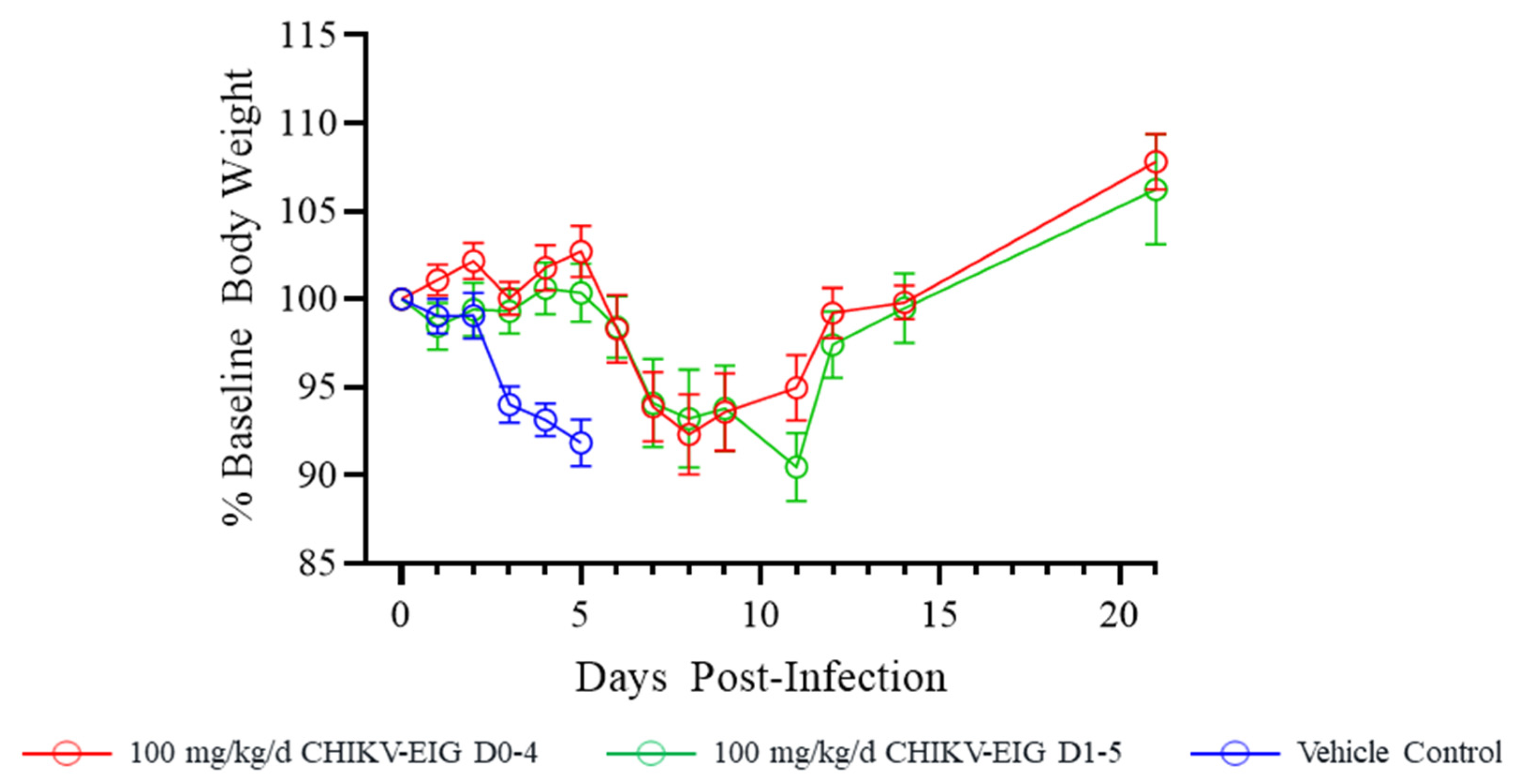
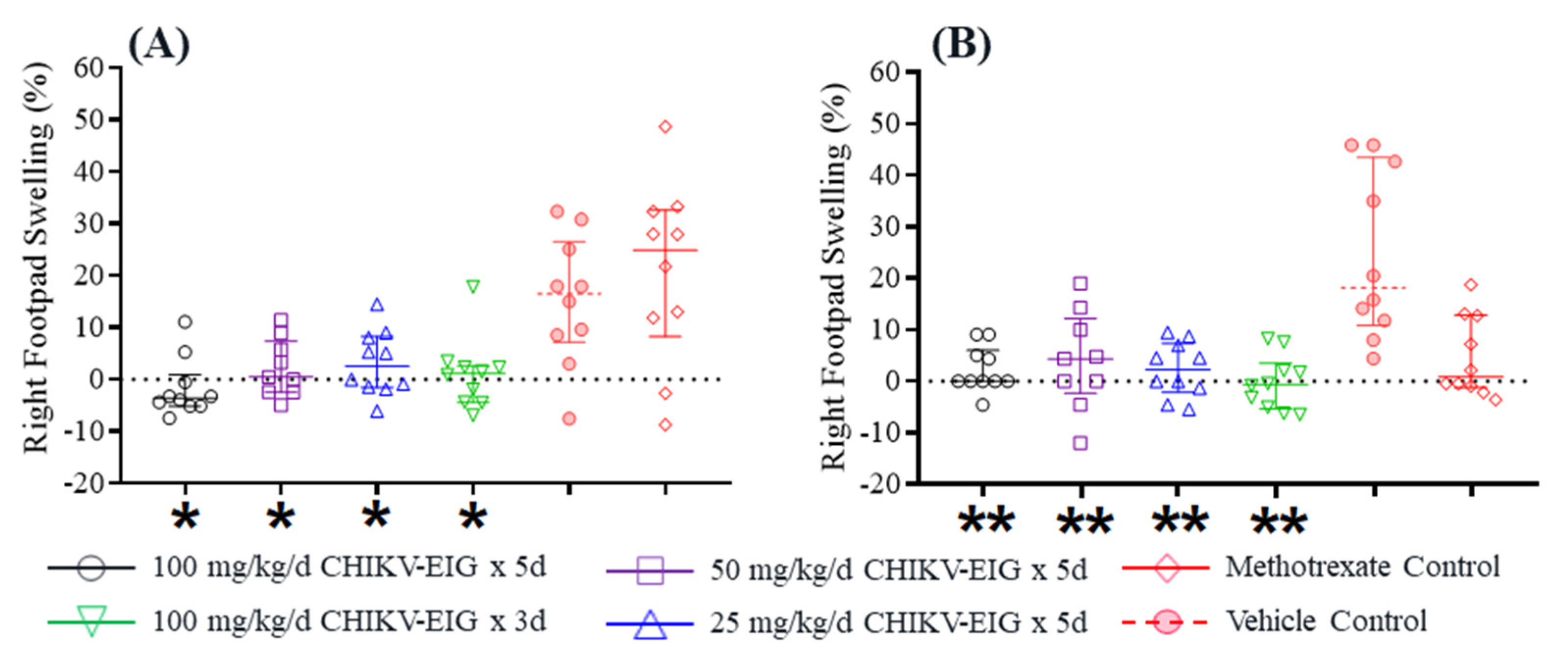
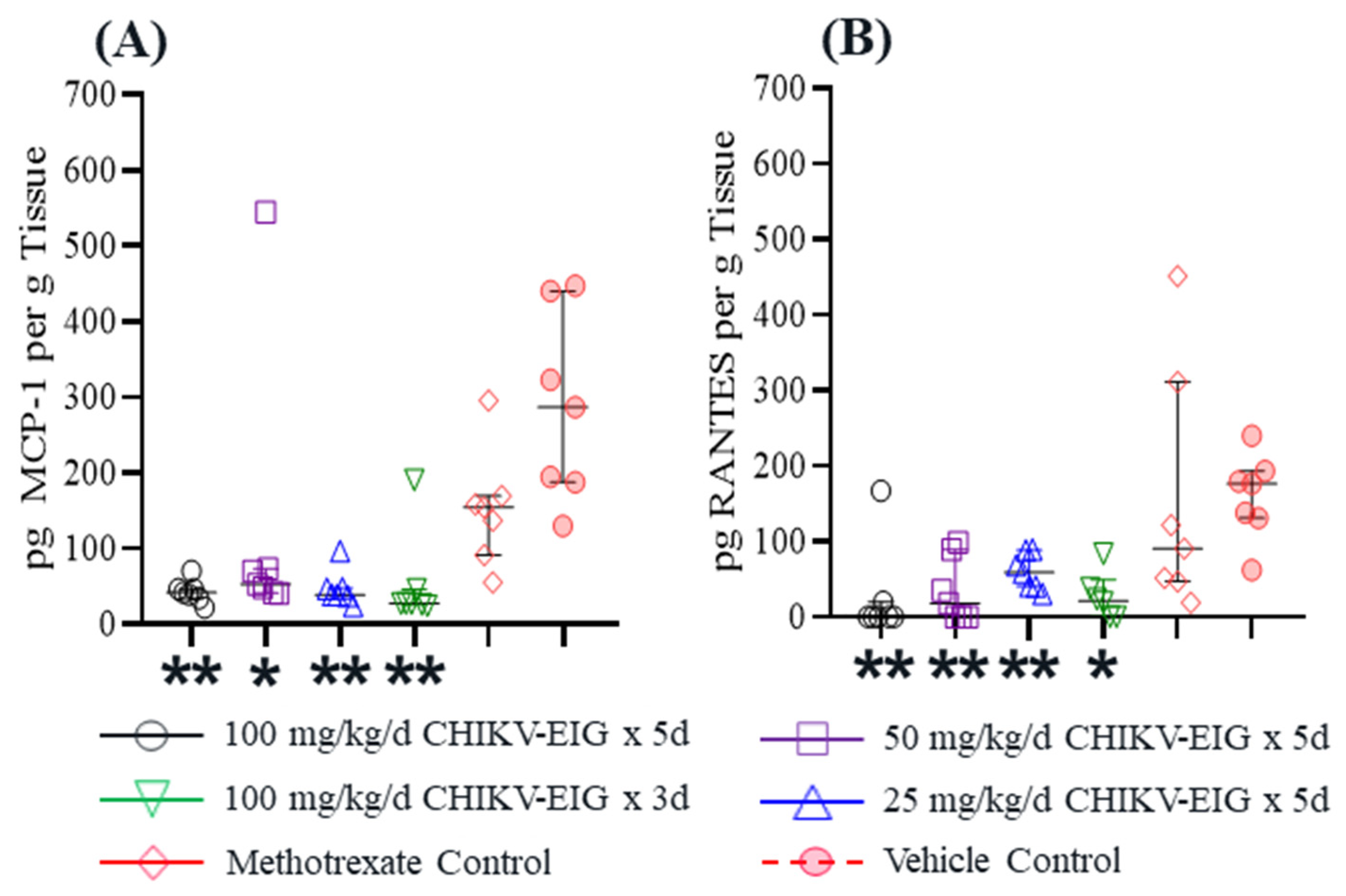
3.4. Protection against Effects of CHIKV in an Immunocompetent Mouse Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef] [PubMed]
- Yactayo, S.; Staples, J.E.; Millot, V.; Cibrelus, L.; Ramo-Pardo, P. Epidemiology of chikungunya in the Americas. J. Infect. Dis. 2016, 214, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Zeller, H.; van Bortel, W.; Sudre, B. Chikungunya: Its history in Africa and Asia and its spread to new regions in 2013–2014. J. Infect. Dis. 2016, 214, S436–S440. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Kenawy, M.A.; Rady, M.H.; Khaled, A.S.; Samy, A.M. Mapping the global potential distributions of two arboviral vectors Aedes aegypti and Ae. Albopictus under changing climate. PLoS ONE 2018, 13, e0210122. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Charlier, C.; Vasilakis, N.; Lecuit, M. Zika, Chikungunya, and Other Emerging Vector-Borne Viral Diseases. Annu. Rev. Med. 2018, 69, 395–408. [Google Scholar] [CrossRef]
- Powers, A.M. How chikungunya virus virology affects its epidemiology and transmission: Implications for influencing public health. J. Infect. Dis. 2016, 214, S449–S452. [Google Scholar] [CrossRef]
- Gasque, P.; Bandjee, M.C.J.; Reyes, M.M.; Viasus, D. Chikungunya pathogenesis: From the clinics to the bench. J. Infect. Dis. 2016, 214, S446–S448. [Google Scholar] [CrossRef]
- Borgherini, G.; Poubeau, P.; Staikowsky, F.; Lory, M.; Le Moullec, N.; Becquart, J.P.; Wengling, C.; Michault, A.; Paganin, F. Outbreak of chikungunya on Reunion Island: Early clinical and laboratory features in 157 adult patients. Clin. Infect. Dis. 2007, 44, 1401–1407. [Google Scholar] [CrossRef]
- Taubitz, W.; Cramer, J.P.; Kapaun, A.; Pfeffer, M.; Drosten, C.; Dobler, G.; Burchard, G.D.; Löscher, T. Chikungunya fever in travelers: Clinical presentation and course. Clin. Infect. Dis. 2007, 45, e1–e4. [Google Scholar] [CrossRef]
- Pialoux, G.; Gauzere, B.A.; Jaureguiberry, S.; Strobel, M. Chikungunya, an epidemic arbovirus. Lancet Infect. Dis. 2007, 7, 319–327. [Google Scholar] [CrossRef]
- Goupil, B.A.; Mores, C.N. A review of chikungunya virus-induced arthralgia: Clinical manifestations, therapeutics, and pathogenesis. Open. Rheumatol. J. 2016, 10, 129–140. [Google Scholar] [CrossRef]
- Sissoko, D.; Malvy, D.; Ezzedine, K.; Renault, P.; Moscetti, F.; Ledrans, M.; Pierre, V. Post-epidemic chikungunya disease on Reunion island: Course of rheumatic manifestations and associated factors over a 15-month period. PLoS Negl. Trop. Dis. 2009, 3, e389. [Google Scholar] [CrossRef]
- Hoarau, J.J.; Bandjee, M.C.J.; Trotot, P.K.; Das, D.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent chronic inflammation and infection by chikungunya arthriogenic alphavirus in spite of a robust host immune response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef]
- Chow, A.; Her, Z.; Ong, E.K.; Chen, J.; Dimatatac, F.; Kwek, D.J.C.; Barkham, T.; Yang, H.; Rénia, L.; Leo, Y.-S.; et al. Persistent arthralgia induced by chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J. Infect. Dis. 2011, 203, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Hawman, D.W.; Stoermer, K.A.; Montgomery, S.A.; Pal, P.; Oko, L.; Diamond, M.S.; Morrison, T.E. Chronic joint disease caused by persistent chikungunya virus infection is controlled by the adaptive immune response. J. Virol. 2013, 87, 13878–13888. [Google Scholar] [CrossRef] [PubMed]
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Bennet, S.R.; McCarty, J.M.; Ramanthan, R.; Mendy, J.; Richardson, J.S.; Smith, J.; Alexander, J.; Ledgerwood, J.E.; de Lame, P.-A.; Royalty Tredo, S.; et al. Safety and immunogenicity of PXVX0317, an aluminium hydroxide-adjuvanted chikungunya virus-like particle vaccine: A randomised, double-blind, parallel-group, phase 2 trial. Lancet Infect. Dis. 2022, 22, 1343–1355. [Google Scholar] [CrossRef]
- Reisinger, E.C.; Tschismarov, R.; Beubler, E.; Widermann, U.; Firbas, C.; Loebermann, M.; Pfeiffer, A.; Mullener, M.; Tauber, M.; Ramsauer, K. Immunogenicity, safety, and tolerability of the measles-vectored chikungunya virus vaccine MV-CHIK: A double-blind, randomised, placebo-controlled and active-controlled phase 2 trial. Lancet 2019, 392, 2718–2727. [Google Scholar] [CrossRef]
- Wressnigg, N.; Hochreiter, R.; Zoihsl, O.; Fritzer, A.; Bézay, N.; Klingler, A.; Lingnau, K.; Schneider, M.; Lundberg, U.; Meinke, A.; et al. Single-shot live-attenuated chikungunya vaccine in healthy adults: A phase 1, randomised controlled trial. Lancet Infect. Dis. 2020, 20, P1193–P1203. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Fink, D.; Hulse, A.; Pratt, R.D. Regulatory considerations in development of vaccines to prevent disease caused by Chikungunya virus. Vaccine 2017, 35, 4851–4858. [Google Scholar] [CrossRef]
- Plante, K.; Wang, E.; Partidos, C.D.; Weger, J.; Gorchakov, R.; Tsetsarkin, K.; Borland, E.M.; Powers, A.M.; Seymour, R.; Stinchcomb, D.T.; et al. Novel chikungunya vaccine candidate with an IRES-based attenuation and host range alteration mechanism. PloS Pathog. 2011, 7, e1002142. [Google Scholar] [CrossRef]
- Roy, C.J.; Adams, A.P.; Wang, E.; Plante, K.; Gorchakov, R.; Seymour, R.L.; Vinet-Oliphant, H.; Weaver, S.C. Chikungunya vaccine candidate is highly attenuated and protects nonhuman primates against telemetrically monitored disease following a single dose. J. Infect. Dis. 2014, 209, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- DeZure, A.D.; Berkowitz, N.M.; Graham, B.S.; Ledgerwood, J.E. Whole-inactivated and virus-like particle vaccine strategies for chikungunya virus. J. Infect. Dis. 2016, 214, S497–S499. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Augiste, A.J.; Kaelber, J.T.; Luo, H.; Rossi, S.L.; Fenton, K.; Leal, G.; Kim, D.Y.; Chiu, W.; Wang, T.; et al. A chikungunya fever vaccine utilizing an insect-specific virus platform. Nat. Med. 2017, 23, 192–199. [Google Scholar] [CrossRef]
- Rossi, S.L.; Comer, J.E.; Wang, E.; Azar, S.R.; Lawrence, W.S.; Plante, J.A.; Ramsauer, K.; Schrauf, S.; Weaver, S.C. Immunogenicity and efficacy of a measles virus-vectored chikungunya vaccine in nonhuman primates. J. Infect. Dis. 2019, 220, 735–742. [Google Scholar] [CrossRef]
- Chen, G.L.; Coates, E.E.; Plummer, S.H.; Carter, C.A.; Berkowitz, N.; Conan-Cibotti, M.; Cox, J.H.; Beck, A.; O’Callahan, M.; Andrews, C.; et al. Efficacy of a chikungunya virus-like particle vaccine on safety and tolerability outcomes: A randomized clinical trial. JAMA 2020, 323, 1369–1377. [Google Scholar] [CrossRef]
- Keller, M.A.; Stiehm, E.R. Passive immunity in prevention and treatment of infectious diseases. Clin. Microbiol. Rev. 2000, 13, 602–614. [Google Scholar] [CrossRef]
- Hopkins, R.J.; Lane, J.M. Clinical efficacy of intramuscular immune globulin: A literature review. Clin. Infect. Dis. 2004, 39, 819–826. [Google Scholar] [CrossRef]
- Shao, H.; McDonald, E.C.; Ginsberg, M.M.; Yee, L.M.; Montgomery, J.R.; Allan-Martinez, F.; Reynolds, M.G.; Tack, D.N.; Davidson, W.; Patel, N.; et al. Secondary and Tertiary Transmission of Vaccinia Virus After Sexual Contact with a Smallpox Vaccinee—San Diego, California, 2012. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 145–147. [Google Scholar]
- Chan, M.; Holtsberg, F.W.; Vu, H.; Howell, K.A.; Leung, A.; van der Hart, E.; Walz, P.H.; Aman, M.J.; Kodihalli, S.; Kobasa, D. Efficacy of ebola glycoprotein-specific equine polyclonal antibody product against lethal ebola virus infection in guinea pigs. J. Infect. Dis. 2018, 218 (Suppl. S5), S603–S611. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wong, G.; Zhao, Y.; Wang, H.; He, S.; Bi, Y.; Chen, W.; Jin, H.; Gai, W.; Chu, D.; et al. Treatment with hyperimmune equine immunoglobulin or immunoglobulin fragments completely protects rodents from ebola virus infection. Sci. Rep. 2016, 6, 24179. [Google Scholar] [CrossRef]
- Kudoyarova-Zubavichene, N.M.; Sergeyev, N.N.; Chepurnov, A.A.; Netesov, S.V. Preparation and use of hyperimmune serum for prophylaxis and therapy of Ebola virus infections. J. Infect. Dis. 1999, 179 (Suppl. S1), S218–S223. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Guo, Z.; Pan, X.; Wang, G.; Zhang, D.; Li, Y.; Tan, B.; Ouyang, L.; Yu, X. Passive immunotherapy for influenza A H5N1 virus infection with equine hyperimmune globulin F(ab’)2 in mice. Respir. Res. 2006, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Branche, E.; Simon, A.Y.; Sheets, N.; Kim, K.; Barker, D.; Nguyen, A.-V.T.; Sahota, H.; Young, M.P.; Salgado, R.; Mamidi, A.; et al. Human polyclonal antibodies prevent lethal zika virus infection in mice. Sci. Rep. 2019, 9, 9857. [Google Scholar] [CrossRef]
- Zaitseva, M.; Kapnick, S.M.; Meseda, C.A.; Shotwell, E.; King, L.R.; Manischewitz, J.; Scott, J.; Kodihalli, S.; Merchlinsky, M.; Nielsen, H.; et al. Passive immunotherapies protect WRvFire and IHD-J-Luc vaccinia virus-infected mice from lethality by reducing viral loads in the upper respiratory tract and internal organs. J. Virol. 2011, 85, 9147–9158. [Google Scholar] [CrossRef]
- Mair-Jenkins, J.; Saavedra-Campos, M.; Baillie, J.K.; Cleary, P.; Khaw, F.; Lim, W.S.; Makki, S.; Rooney, K.D.; Convalescent Plasma Study Group; Nguyen-Van-Tham, J.S.; et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: A systematic review and exploratory meta-analysis. J. Infect. Dis. 2015, 211, 80–90. [Google Scholar] [CrossRef]
- Jha, A.; Barker, D.; Lew, J.; Manoharan, V.; van Kessel, J.; Haupt, R.; Toth, D.; Frieman, M.; Falzarano, R.; Kodihalli, S. Efficacy of COVID-HIGIV in animal models of SARS-CoV-2 infection. Sci. Rep. 2022, 12, 16956. [Google Scholar] [CrossRef]
- Barbier, M.; Lee, K.S.; Vikharankar, M.S.; Rajpathak, S.N.; Kadam, N.; Wong, T.Y.; Russ, B.P.; Cyphert, H.A.; Miller, O.A.; Rader, D.A.; et al. Passive immunization with equine RBD-specific Fab protects K18-hACE2-mice against Alpha or Beta variants of SARS-CoV-2. Front. Immunol. 2022, 11, 948431. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Maguiraga, L.; Noret, M.; Brun, S.; Le Grand, R.; Gras, G.; Roques, P. Chikungunya disease: Infection-associated markers from the acute to the chronic phase of arbovirus-induced arthralgia. PLoS Negl. Trop. Dis. 2012, 6, e1446. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Smith, S.A.; Silva, L.A.; Fox, J.M.; Flyak, A.I.; Krose, N.; Sapparapu, G.; Khomandiak, S.; Askhbrook, A.W.; Kahle, K.M.; Fong, R.H.; et al. Isolation and characterization of broad and ultrapotent human monoclonal antibodies with therapeutic activity against chikungunya virus. Cell Host Microbe 2015, 18, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Long, F.; Edeling, M.A.; Lin, H.; van Duijl-Richter, M.K.S.; Fong, R.H.; Kahle, K.M.; Smit, J.M.; Jin, J.; Simmons, G.; et al. Broadly neutralizing alphavirus antibodies bind an epitope on e2 and inhibit entry and egress. Cell 2015, 163, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Simmons, G. Antiviral function of monoclonal antibodies against Chikungunya virus. Viruses 2019, 11, 305. [Google Scholar] [CrossRef] [PubMed]
- Hucke, F.I.L.; Bestehorn-Willmann, M.; Bugert, J.J. Prophylactic strategies to control chikungunya virus infection. Virus Genes 2021, 57, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Henderson Sousa, F.; Ghaisani Komarudin, A.; Findlay-Greene, F.; Bowolaksono, A.; Sasmono, R.T.; Stevens, C.; Barlow, P.G. Evolution and immunopathology of chikungunya virus informs therapeutic development. Dis. Model. Mech. 2023, 16, dmm049804. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Kam, Y.-W.; Fric, J.; Malleret, B.; Koh, E.G.L.; Prakash, C.; Huang, W.; Lee, W.W.L.; Lin, C.; Lin, R.T.P.; et al. Chikungunya virus neutralization antigens and direct cell-to-cell transmission are revealed by human antibody-escape mutants. PLoS Pathog. 2011, 7, e1002390. [Google Scholar] [CrossRef]
- Parrera, G.S.; Astacio, H.; Tunga, P.; Anderson, D.M.; Hall, C.L.; Richardson, J.S. Use of Botulism Antitoxin Heptavalent (A, B, C, D, E, F, G)–(Equine) (BAT®) in clinical study subjects and patients: A 15-year systematic safety review. Toxins 2021, 14, 19. [Google Scholar] [CrossRef]
- Richardson, J.S.; Parrera, G.S.; Astacio, H.; Sahota, H.; Anderson, D.M.; Hall, C.; Babinchak, T. Safety and Clinical Outcomes of an Equine-derived Heptavalent Botulinum Antitoxin Treatment for Confirmed or Suspected Botulism in the United States. Clin. Infect. Dis. 2020, 70, 1950–1957. [Google Scholar] [CrossRef]
- Wilde, H.; Khawplod, P.; Hemachdha, T.; Sitprija, V. Postexposure treatment of rabies infection: Can it be done without immunoglobulin? Clin. Infect. Dis. 2002, 34, 477–480. [Google Scholar] [CrossRef]
- Naiditch, M.J.; Bower, A.G. Diphtheria a study of 1433 cases observed during a ten year period at the Los Angeles County Hospital. Am. J. Med. 1954, 17, 229–245. [Google Scholar] [CrossRef]
- Bampoe, V.D.; Boswell, H.C.; Yu, Y.C.; Acosta, A.M. A review of adverse events from the use of diphtheria antitoxin (DAT) in the United States, 2004–2019. Clin. Infect. Dis. 2022, 74, 2082–2083. [Google Scholar] [CrossRef]
- USDA. Animal Welfare Act and Animal Welfare Regulations. Available online: https://www.aphis.usda.gov/animal_welfare/downloads/AC_BlueBook_AWA_508_comp_version.pdf (accessed on 19 October 2022).
- National Research Council of the National Academies. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011; ISBN 10-0-309-15400-6. [Google Scholar]
- Adam, A.; Luo, H.; Osman, S.R.; Wang, B.; Roundy, C.M.; Auguste, A.J.; Plante, K.S.; Peng, B.; Thangamani, S.; Frolova, E.I.; et al. Optimized production and immuongenicity of an insect virus-based chikungunya virus candidate vaccine in cell culture and animal models. Emerg. Microbes Infect. 2021, 10, 305–316. [Google Scholar] [CrossRef]
- Kodihalli, S.; Emanuel, A.; Takla, T.; Hua, Y.; Hobbs, C.; LeClaire, R.; O’Donnell, D.C. Therapeutic efficacy of equine botulism antitoxin in Rhesus macaques. PLoS ONE 2017, 12, e0186892. [Google Scholar] [CrossRef] [PubMed]
- Nasar, F.; Palacios, G.; Gorchakov, R.V.; Guzman, H.; Travassos Da Rosa, A.P.; Savji, N.; Popov, V.L.; Sherman, M.B.; Lipkin, I.; Tesh, R.B.; et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 14622–14627. [Google Scholar] [CrossRef] [PubMed]
- Levitt, N.H.; Ramsburg, H.H.; Hasty, S.E.; Repik, P.M.; Cole, F.E., Jr.; Lupton, H.W. Development of an attenuated strain of Chikungunya virus for use in vaccine production. Vaccine 1986, 4, 157–162. [Google Scholar] [CrossRef]
- Beaty, B.J.; Calisher, C.H.; Shope, R.E. Arboviruses. In Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infections; Schmidt, N.J., Emmons, R.W., Eds.; American Public Health Association: Washington, DC, USA, 1989; pp. 797–855. [Google Scholar]
- Julander, J.G.; Dagley, A.; Gebre, M.; Komeno, T.; Makajima, N.; Smee, D.F.; Furuta, Y. Strain-dependent disease and response to favipiravir treatment in mice infected with Chikungunya virus. Antivir. Res. 2020, 182, 104904. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, C.H. A simple method of estimating fifty percent endpoint. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Dagley, A.; Ennis, J.; Turner, J.D.; Rood, K.A.; van Wettere, A.J.; Gowen, B.B.; Julander, J.G. Protection against Chikungunya virus induced arthralgia following prophylactic treatment with adenovirus vectored interferon (mDEF201). Antivir. Res. 2014, 108, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient Type-I Interferon signalling are risk factors for severe disease. PloS. Pathog. 2008, 4, e29. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.L.; Burke, C.W.; Higgs, S.T.; Klimstra, W.B.; Ryman, K.D. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type chikungunya virus but not with the cell culture-adapted live attenuated 181/25 vaccine candidate. Virology 2012, 452, 103–112. [Google Scholar] [CrossRef]
- Pal, P.; Dowd, K.A.; Brien, J.D.; Edeling, M.A.; Gorlatov, S.; Johnson, S.; Lee, I.; Akahata, W.; Nabel, G.J.; Richter, M.K.S.; et al. Development of a highly protective combination monoclonal antibody therapy against chikungunya virus. PloS. Pathog. 2013, 9, e1003312. [Google Scholar] [CrossRef]
- Partidos, C.D.; Wegner, J.; Brewoo, J.; Seymour, R.; Borland, E.M.; Ledermann, J.P.; Powers, A.M.; Weaver, S.C.; Stinchcomb, D.T.; Osorio, J.E. Probing the attenuation and protective efficacy of a candidate chikungunya virus vaccine in mice with compromised interferon (IFN) signaling. Vaccine 2011, 29, 3067–3073. [Google Scholar] [CrossRef]
- Couderc, T.; Khandoudi, N.; Grandadam, M.; Visse, C.; Gangneux, N.; Bagot, S.; Prost, J.; Lecuit, M. Prophylaxis and therapy for Chikungunya virus. J. Infect. Dis. 2009, 200, 516–523. [Google Scholar] [CrossRef]
- Plante, K.S.; Rossi, S.L.; Bergren, N.A.; Seymour, R.L.; Weaver, S.C. Extended preclinical safety, efficacy and stability testing of a live-attenuated chikungunya vaccine candidate. PLoS Negl. Trop. Dis. 2015, 9, e0004007. [Google Scholar] [CrossRef] [PubMed]
- Dagley, A.; Julander, J.G. A.; Julander, J.G. A mouse model of Chikungunya virus with utility in antiviral studies. In Antiviral Methods and Protocols; Methods in Molecular Biology (Methods and Protocols); Gong, E., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 1030, pp. 439–448. [Google Scholar] [CrossRef]
- Lum, F.M.; Teo, T.H.; Lee, W.W.L.; Kam, Y.-W.; Rénia, L.; Ng, L.F.P. An essential role of antibodies in the control of Chikungunya virus infection. J. Immunol. 2013, 90, 6295–6302. [Google Scholar] [CrossRef] [PubMed]
- Yoon, I.K.; Alera, M.T.; Lago, C.B.; Tac-An, I.A.; Villa, D.; Fernandez, S.; Thaisomboonsuk, B.; Klungthong, C.; Levy, J.W.; Velasco, J.M.; et al. High rate of subclinical Chikungunya virus infection and association of neutralizing antibody with protection in a prospective cohort in the Philippines. PLoS Negl. Trop. Dis. 2015, 9, e0003764. [Google Scholar] [CrossRef]
- Jin, J.; Liss, N.M.; Chen, D.H.; Liao, M.; Fox, J.M.; Shimak, R.M.; Fong, R.H.; Chafets, D.; Bakkour, S.; Keating, S.; et al. Neutralizing monoclonal antibodies block Chikungunya virus entry and release by targeting an epitope critical to viral pathogenesis. Cell Rep. 2015, 13, 2553–2564. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Fox, J.M.; Hawman, D.W.; Huang, Y.S.; Messaoudi, I.; Kreklywich, C.; Denton, M.; Legasse, A.W.; Smith, P.P.; Johnson, S.; et al. Chikungunya viruses that escape monoclonal antibody therapy are clinically attenuated, stable, and not purified in mosquitoes. J. Virol. 2014, 88, 8213–8226. [Google Scholar] [CrossRef]
- Porta, J.; Mangala Prasad, V.; Wang, C.I.; Akahata, W.; Ng, L.F.; Rossmann, M.G. Structural studies of Chikungunya virus-like particles complexed with human antibodies: Neutralization and cell-to-cell transmission. J. Virol. 2016, 90, 1169–1177. [Google Scholar] [CrossRef]
- Haese, N.N.; Broeckel, R.M.; Hawman, D.W.; Heise, M.T.; Morrison, T.E.; Streblow, D.N. Animal models of Chikungunya virus infection and disease. J. Infect. Dis. 2016, 214, S482–S487. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Jochmans, D.; Verbeken, E.; Neyts, J.; Delang, L. Antiviral treatment efficiently inhibits chikungunya virus infection in the joints of mice during the acute but not during the chronic phase of the infection. Antivir. Res. 2018, 149, 113–117. [Google Scholar] [CrossRef]
- Amaral, J.K.; Taylor, P.C.; Teixeira, M.M.; Morrison, T.E.; Schoen, R.T. The clinical features, pathogenesis and methotrexate therapy of chronic chikungunya arthritis. Viruses 2019, 11, 289. [Google Scholar] [CrossRef]
- Tun, Y.M.; Charunwatthana, P.; Duangdee, C.; Satayarak, J.; Suthisawat, S.; Likhit, O.; Lakhotia, D.; Kosoltanapiwat, N.; Sukphopetch, P.; Boonnak, K. Virological, serological and clinical analysis of chikungunya virus infection in Thai patients. Viruses 2022, 14, 1805. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Nayak, K.; Tanwar, N.; Gaind, R.; Gupta, B.; Shastri, J.S.; Bhatanagar, R.K.; Kaja, M.K.; Chandele, A.; Sunil, S. Clinical, serological, and virological analysis of 572 chikungunya patients from 2010 to 2013 in India. Clin. Infect. Dis. 2017, 65, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Chow, A.; Sun, Y.J.; Kwek, D.J.C.; Lim, P.; Dimatatac, F.; Ng, L.; Ooi, E.; Choo, K.; Her, Z.; et al. IL-1beta, IL-6, and RANTES as biomarkers of Chikungunya severity. PLoS ONE 2009, 4, e4261. [Google Scholar] [CrossRef]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Schall, T.J.; Bacon, K.; Toy, K.J.; Goeddel, D.V. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 1990, 347, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Ajuebor, M.N.; Hogaboam, C.M.; Kunkel, S.L.; Proudfoot, A.E.I.; Wallace, J.L. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J. Immunol. 2001, 166, 552–558. [Google Scholar] [CrossRef]
- Patel, D.D.; Zachariah, J.P.; Whichard, L.P. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin. Immunol. 2001, 98, 39–45. [Google Scholar] [CrossRef]
- Agere, S.A.; Akhtar, N.; Watson, J.M.; Ahmed, S. RANTES/CCL5 Induces Collagen Degradation by Activating MMP-1 and MMP-13 Expression in Human Rheumatoid Arthritis Synovial Fibroblasts. Front. Immunol. 2017, 8, 1341. [Google Scholar] [CrossRef]
- Katschke, K.J.; Rottman, J.B.; Ruth, J.H.; Qin, S.; Wu, L.; LaRosa, G.; Ponath, P.; Park, C.C.; Pope, R.M.; Koch, A.E. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001, 44, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Pierer, M.; Rethage, J.; Seibl, R.; Lauener, R.; Brentano, F.; Wagner, U.; Hantzschel, H.; Michel, B.A.; Gay, R.E.; Gay, S.; et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J. Immunol. 2004, 172, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.; Joyner, M.J. The principles of antibody therapy for infectious diseases with relevance for COVID-19. mBio 2021, 12, e03372-20. [Google Scholar] [CrossRef]
- Tharmalingam, T.; Han, X.; Wozniak, A.; Saward, L. Polyclonal hyper immunoglobulin: A proven treatment and prophylaxis platform for passive immunization to address existing and emerging diseases. Hum. Vaccines Immunother. 2021, 18, 1886560. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.M.; Jones, T.J.; Barr, K.L. Antibodies for Venezuelan Equine Encephalitis Virus protect embryoid bodies from Chikungunya virus. Viruses 2020, 12, 262. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Lineage | Titer (PRNT80) |
|---|---|---|
| CHIKV 181/25 | - | 5120 |
| 99,659 | Asian | 5120 |
| LR | Indian Ocean | 2560 |
| 37,997 | West African | 5120 |
| Virus | Strain | Titer (PRNT80) |
|---|---|---|
| O’Nyong-Nyong | SG650 | 640 |
| Semliki Forest Virus | Kumba | 40 |
| Mayaro Virus | FMD3212 | <20 |
| TRVL15537 | 20 | |
| BeAR505411 | <20 | |
| INHRR11a-10 | 20 | |
| Ross River Virus | T48 | <20 |
| Treatment Group | Mean Peak Percent Weight Loss (95% CI) | Median Time to Peak Weight Loss in Days (95% CI) | Sidak-Adjusted Log-Rank Test vs. Vehicle Control |
|---|---|---|---|
| Vehicle Control | 8.17 (5.56, 10.78) | 5.00 (3.00, 5.00) | n/a |
| CHIKV-EIG 100 mg/kg/day (qd X5, i.p. beginning 5 h post-infection) | 7.70 (3.19, 12.20) | 8.00 (7.00, 9.00) | 0.0057 * |
| CHIKV-EIG 100 mg/kg/day (qd X5, i.p. beginning day 1 post-infection) | 9.53 (5.71, 13.34) | 11.0 (4.00, 11.0) | <0.001 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barker, D.; Han, X.; Wang, E.; Dagley, A.; Anderson, D.M.; Jha, A.; Weaver, S.C.; Julander, J.; Nykiforuk, C.; Kodihalli, S. Equine Polyclonal Antibodies Prevent Acute Chikungunya Virus Infection in Mice. Viruses 2023, 15, 1479. https://doi.org/10.3390/v15071479
Barker D, Han X, Wang E, Dagley A, Anderson DM, Jha A, Weaver SC, Julander J, Nykiforuk C, Kodihalli S. Equine Polyclonal Antibodies Prevent Acute Chikungunya Virus Infection in Mice. Viruses. 2023; 15(7):1479. https://doi.org/10.3390/v15071479
Chicago/Turabian StyleBarker, Douglas, Xiaobing Han, Eryu Wang, Ashley Dagley, Deborah M. Anderson, Aruni Jha, Scott C. Weaver, Justin Julander, Cory Nykiforuk, and Shantha Kodihalli. 2023. "Equine Polyclonal Antibodies Prevent Acute Chikungunya Virus Infection in Mice" Viruses 15, no. 7: 1479. https://doi.org/10.3390/v15071479
APA StyleBarker, D., Han, X., Wang, E., Dagley, A., Anderson, D. M., Jha, A., Weaver, S. C., Julander, J., Nykiforuk, C., & Kodihalli, S. (2023). Equine Polyclonal Antibodies Prevent Acute Chikungunya Virus Infection in Mice. Viruses, 15(7), 1479. https://doi.org/10.3390/v15071479