
IGF2BP1—An Oncofetal RNA-Binding Protein Fuels Tumor Virus Propagation



Abstract
1. Introduction
2. Materials and Methods
3. Human Oncogenic Viruses
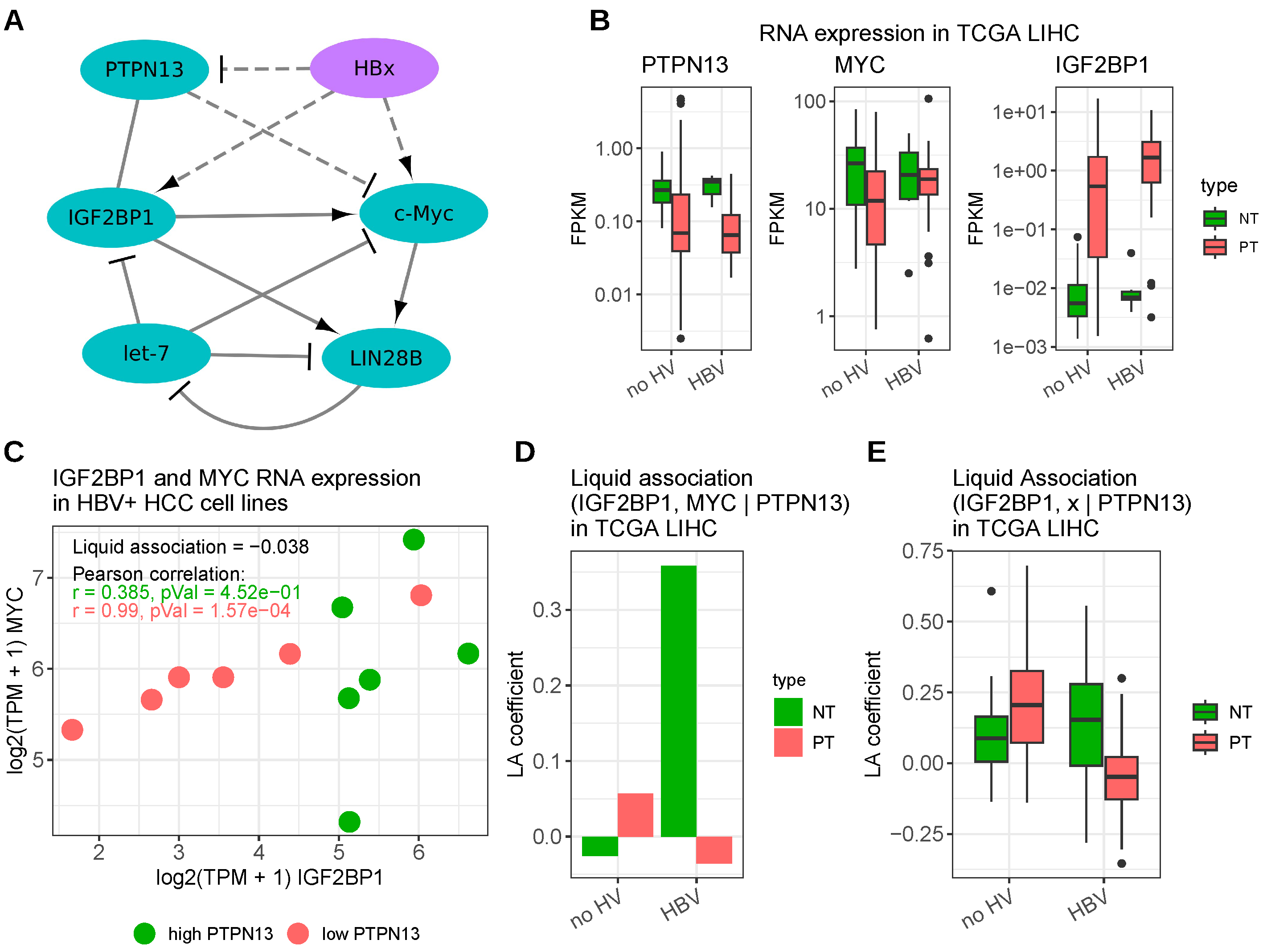
3.1. Hepatitis B Virus

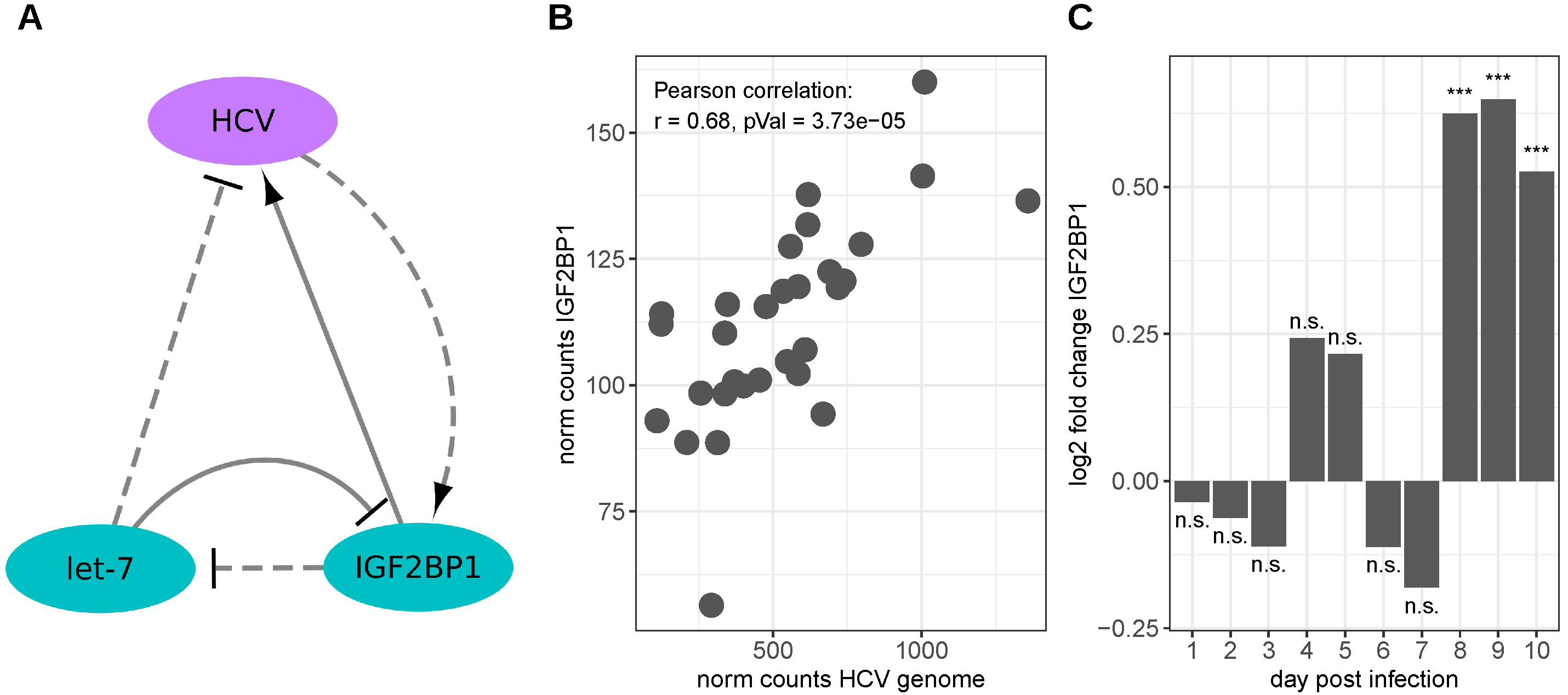
3.2. Hepatitis C Virus
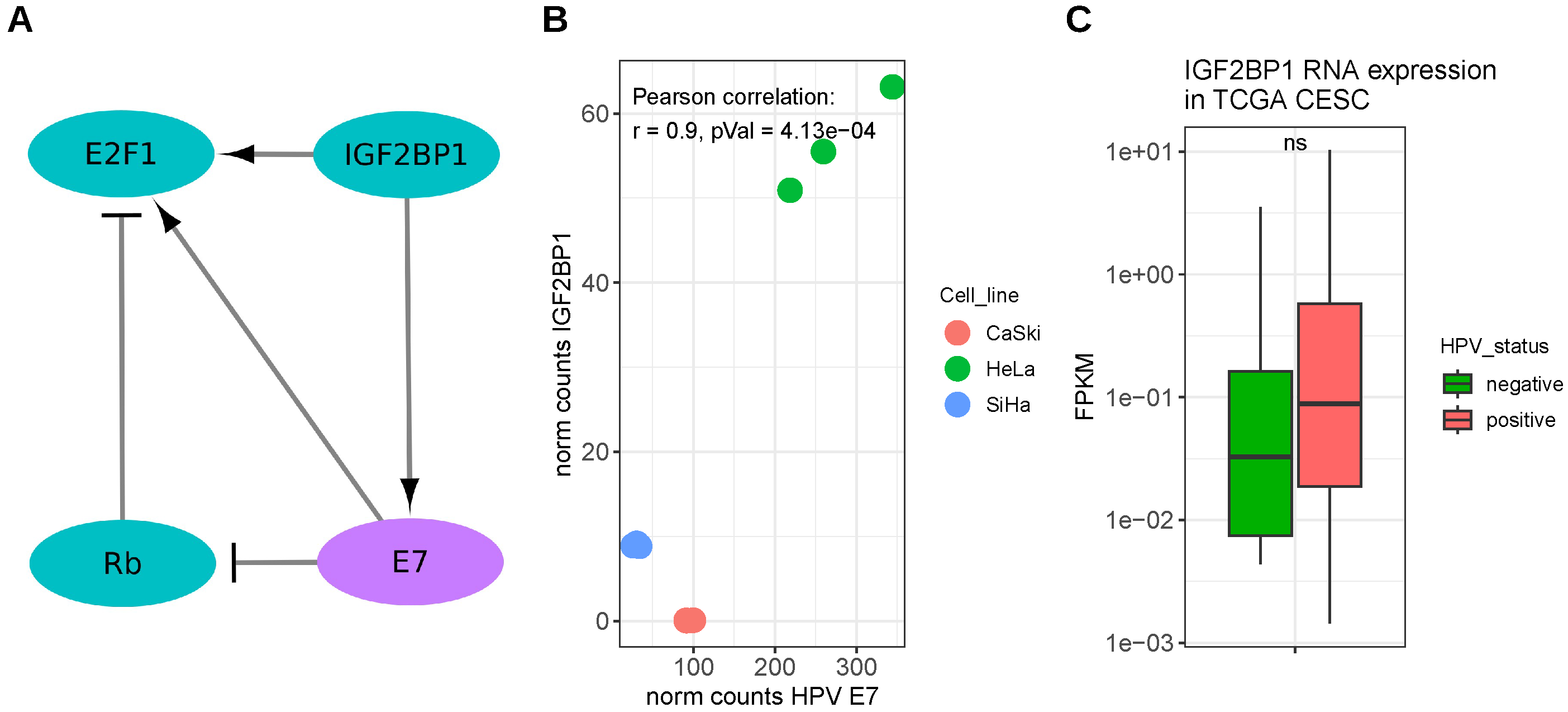
3.3. Human Papillomavirus
4. Human Non-Oncogenic Viruses
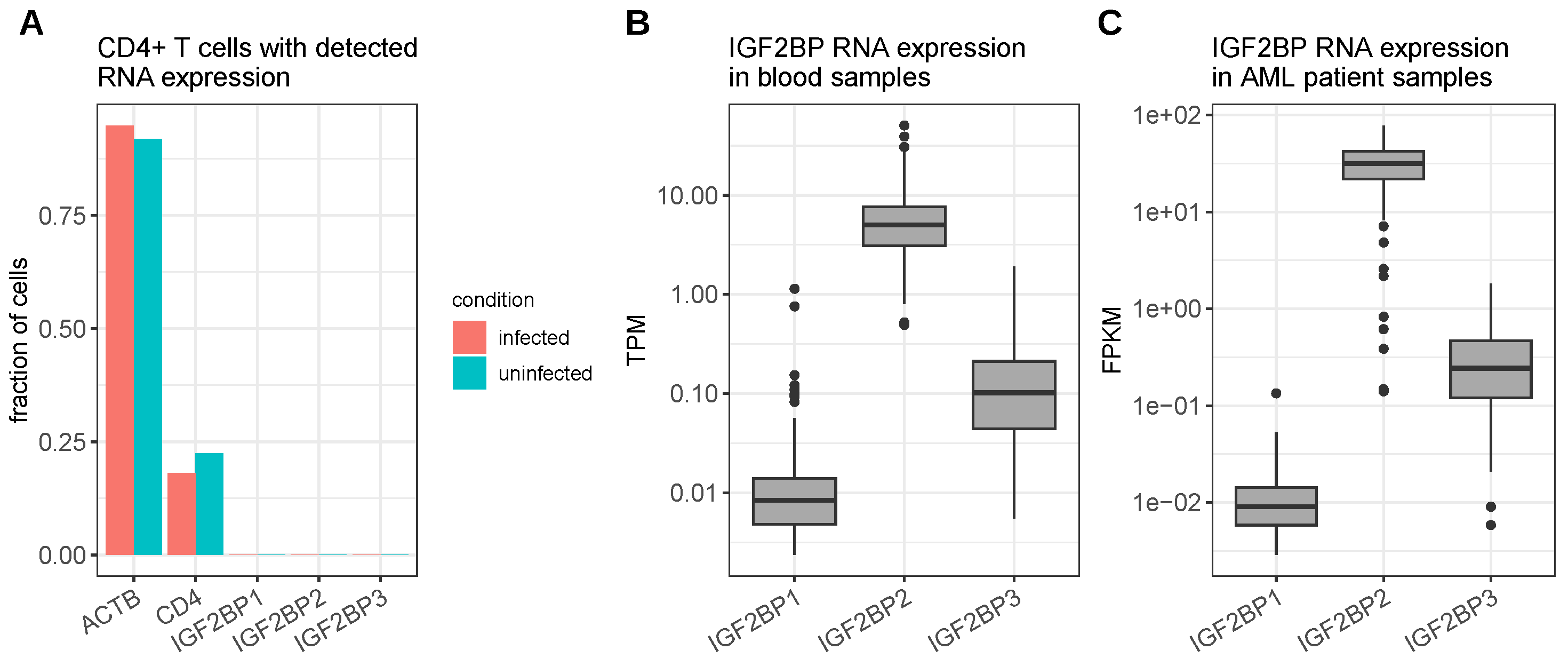
4.1. Human Immunodeficiency Virus Type 1
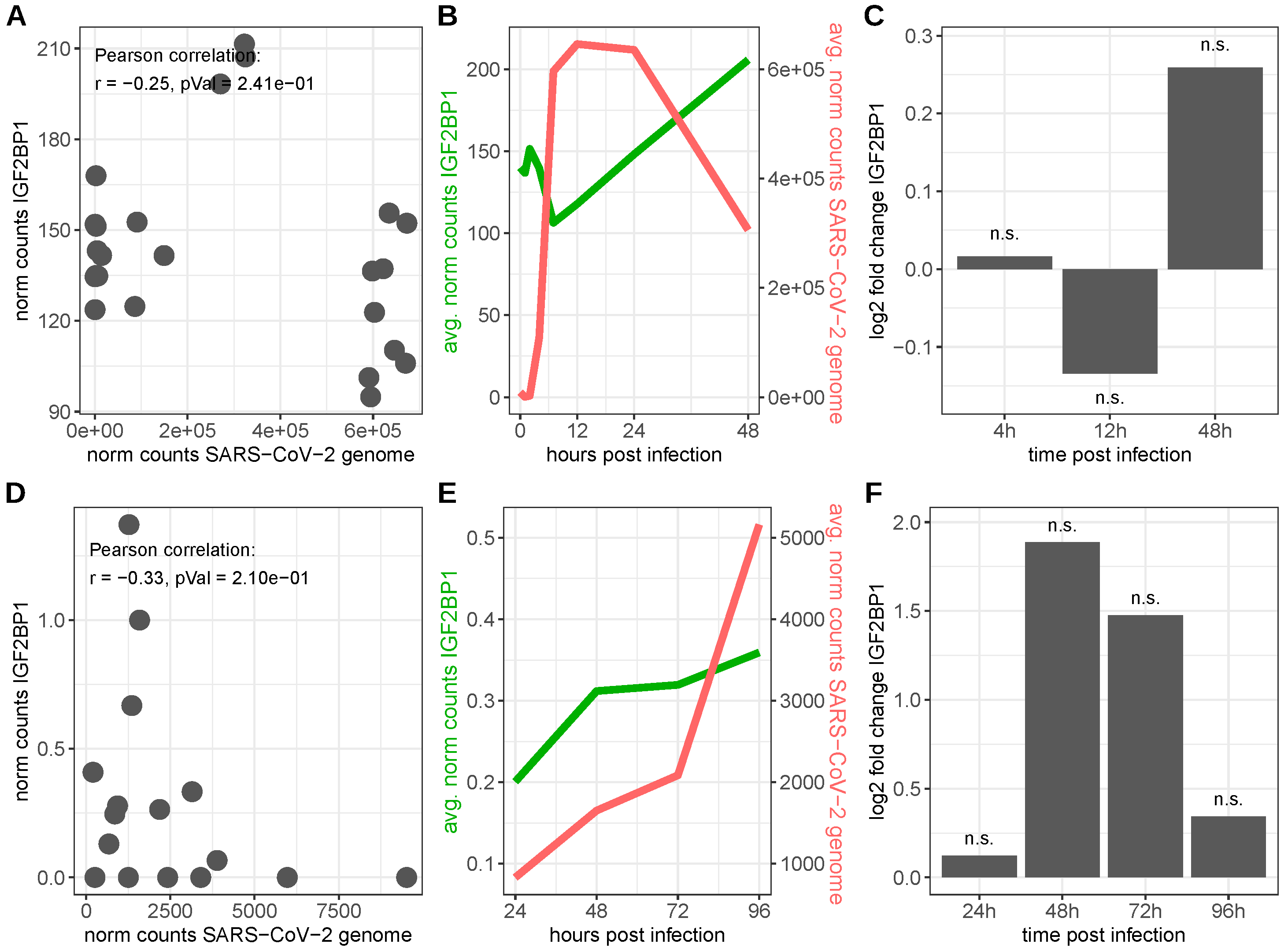
4.2. Severe Acute Respiratory Syndrome Coronavirus 2
4.3. Other Human Viruses
5. Non-Human Viruses
6. Discussion
7. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nagy, P.D.; Pogany, J. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 2011, 10, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Purdy, J.G.; Luftig, M.A. Reprogramming of cellular metabolic pathways by human oncogenic viruses. Curr. Opin. Virol. 2019, 39, 60–69. [Google Scholar] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef]
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [PubMed]
- Cook, L. Polyomaviruses. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Nevels, M.; Täuber, B.; Spruss, T.; Wolf, H.; Dobner, T. “Hit-and-run” transformation by adenovirus oncogenes. J. Virol. 2001, 75, 3089–3094. [Google Scholar]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Dass, D.; Dhotre, K.; Chakraborty, M.; Nath, A.; Banerjee, A.; Bagchi, P.; Mukherjee, A. MiRNAs in Herpesvirus infection: Powerful regulators in small packages. Viruses 2023, 15, 429. [Google Scholar] [CrossRef]
- Megahed, F.; Tabll, A.; Atta, S.; Ragheb, A.; Smolic, R.; Petrovic, A.; Smolic, M. MicroRNAs: Small molecules with significant functions, particularly in the context of viral hepatitis B and C infection. Medicina 2023, 59, 173. [Google Scholar]
- Alshahrani, S.H.; Alameri, A.A.; Kahar, F.; Alexis Ramírez-Coronel, A.; Fadhel Obaid, R.; Alsaikhan, F.; Zabibah, R.S.; Qasim, Q.A.; Altalbawy, F.M.A.; Fakri Mustafa, Y.; et al. Overview of the role and action mechanism of microRNA-128 in viral infections. Microb. Pathog. 2023, 176, 106020. [Google Scholar] [CrossRef]
- Rashid, F.; Zaongo, S.D.; Song, F.; Chen, Y. The diverse roles of miRNAs in HIV pathogenesis: Current understanding and future perspectives. Front. Immunol. 2022, 13, 1091543. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, Y.P.; Li, K.; Ye, Q.; Zhou, H.Y.; Sun, H.; Li, X.; Yu, L.; Deng, Y.Q.; Li, R.T.; et al. A methylome of SARS-CoV-2 in host cells. Cell Res. 2021, 31, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Lichinchi, G.; Zhao, B.S.; Wu, Y.; Lu, Z.; Qin, Y.; He, C.; Rana, T.M. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe 2016, 20, 666–673. [Google Scholar] [CrossRef]
- Tirumuru, N.; Zhao, B.S.; Lu, W.; Lu, Z.; He, C.; Wu, L. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife 2016, 5, e15528. [Google Scholar] [CrossRef]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G.M.; Bansal, V.; Wang, Y.; Mason, C.E.; Rana, T.M. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016, 1, 16011. [Google Scholar] [CrossRef]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef]
- Tan, B.; Gao, S.J. RNA epitranscriptomics: Regulation of infection of RNA and DNA viruses by N6-methyladenosine (m6A)). Rev. Med. Virol. 2018, 28, e1983. [Google Scholar] [CrossRef]
- Ioannidis, P.; Mahaira, L.; Papadopoulou, A.; Teixeira, M.R.; Heim, S.; Andersen, J.A.; Evangelou, E.; Dafni, U.; Pandis, N.; Trangas, T. CRD-BP: A c-Myc mRNA stabilizing protein with an oncofetal pattern of expression. Anticancer Res. 2003, 23, 2179–2183. [Google Scholar]
- Degrauwe, N.; Suvà, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef]
- Hattori, A.; Buac, K.; Ito, T. Regulation of Stem Cell Self-Renewal and Oncogenesis by RNA-Binding Proteins. Adv. Exp. Med. Biol. 2016, 907, 153–188. [Google Scholar] [PubMed]
- Bell, J.L.; Wächter, K.; Mühleck, B.; Pazaitis, N.; Köhn, M.; Lederer, M.; Hüttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell. Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, P.; Kottaridi, C.; Dimitriadis, E.; Courtis, N.; Mahaira, L.; Talieri, M.; Giannopoulos, A.; Iliadis, K.; Papaioannou, D.; Nasioulas, G.; et al. Expression of the RNA-binding protein CRD-BP in brain and non-small cell lung tumors. Cancer Lett. 2004, 209, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Köbel, M.; Weidensdorfer, D.; Reinke, C.; Lederer, M.; Schmitt, W.D.; Zeng, K.; Thomssen, C.; Hauptmann, S.; Hüttelmaier, S. Expression of the RNA-binding protein IMP1 correlates with poor prognosis in ovarian carcinoma. Oncogene 2007, 26, 7584–7589. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Bley, N.; Glaß, M.; Busch, B.; Rousseau, V.; Misiak, D.; Fuchs, T.; Lederer, M.; Hüttelmaier, S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018, 46, 6285–6303. [Google Scholar] [CrossRef]
- Glaß, M.; Michl, P.; Hüttelmaier, S. RNA Binding Proteins as Drivers and Therapeutic Target Candidates in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 4190. [Google Scholar] [CrossRef]
- Gutschner, T.; Hämmerle, M.; Pazaitis, N.; Bley, N.; Fiskin, E.; Uckelmann, H.; Heim, A.; Groß, M.; Hofmann, N.; Geffers, R.; et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology 2014, 59, 1900–1911. [Google Scholar] [CrossRef]
- Bell, J.L.; Turlapati, R.; Liu, T.; Schulte, J.H.; Hüttelmaier, S. IGF2BP1 harbors prognostic significance by gene gain and diverse expression in neuroblastoma. J. Clin. Oncol. 2015, 33, 1285–1293. [Google Scholar] [CrossRef]
- Li, S.; Jiang, M. Elevated insulin-like growth factor 2 mRNA binding protein 1 levels predict a poor prognosis in patients with breast carcinoma using an integrated multi-omics data analysis. Front. Genet. 2022, 13, 994003. [Google Scholar] [CrossRef]
- Müller, S.; Glaß, M.; Singh, A.K.; Haase, J.; Bley, N.; Fuchs, T.; Lederer, M.; Dahl, A.; Huang, H.; Chen, J.; et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019, 47, 375–390. [Google Scholar] [CrossRef]
- Busch, B.; Bley, N.; Müller, S.; Glaß, M.; Misiak, D.; Lederer, M.; Vetter, M.; Strauß, H.G.; Thomssen, C.; Hüttelmaier, S. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016, 44, 3845–3864. [Google Scholar] [CrossRef] [PubMed]
- Leeds, P.; Kren, B.T.; Boylan, J.M.; Betz, N.A.; Steer, C.J.; Gruppuso, P.A.; Ross, J. Developmental regulation of CRD-BP, an RNA-binding protein that stabilizes c-myc mRNA in vitro. Oncogene 1997, 14, 1279–1286. [Google Scholar] [CrossRef]
- Boyerinas, B.; Park, S.M.; Murmann, A.E.; Gwin, K.; Montag, A.G.; Zillhardt, M.; Hua, Y.J.; Lengyel, E.; Peter, M.E. Let-7 modulates acquired resistance of ovarian cancer to Taxanes via IMP-1-mediated stabilization of multidrug resistance 1. Int. J. Cancer 2012, 130, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Elcheva, I.; Goswami, S.; Noubissi, F.K.; Spiegelman, V.S. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 2009, 35, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Glaß, M.; Misiak, D.; Bley, N.; Müller, S.; Hagemann, S.; Busch, B.; Rausch, A.; Hüttelmaier, S. IGF2BP1, a Conserved Regulator of RNA Turnover in Cancer. Front. Mol. Biosci. 2021, 8, 632219. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Bley, N.; Busch, B.; Glaß, M.; Lederer, M.; Misiak, C.; Fuchs, T.; Wedler, A.; Haase, J.; Bertoldo, J.B.; et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020, 48, 8576–8590. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef]
- Lu, H.; Li, W.; Noble, W.S.; Payan, D.; Anderson, D.C. Riboproteomics of the hepatitis C virus internal ribosomal entry site. J. Proteome Res. 2004, 3, 949–957. [Google Scholar] [CrossRef]
- Wang, L.; Zhan, G.; Maimaitiyiming, Y.; Su, Y.; Lin, S.; Liu, J.; Su, K.; Lin, J.; Shen, S.; He, W.; et al. m6A modification confers thermal vulnerability to HPV E7 oncotranscripts via reverse regulation of its reader protein IGF2BP1 upon heat stress. Cell Rep. 2022, 41, 111546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.; Ren, L.; Ju, X.; Gong, M.; Rao, J.; Sun, L.; Li, P.; Ding, Q.; Wang, J.; et al. Comparison of viral RNA-host protein interactomes across pathogenic RNA viruses informs rapid antiviral drug discovery for SARS-CoV-2. Cell Res. 2022, 32, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, S.; Hüttelmaier, S.; Schierhorn, A.; Behrens, S.E.; Ostareck-Lederer, A.; Ostareck, D.H. IGF2BP1 enhances HCV IRES-mediated translation initiation via the 3’UTR. RNA 2009, 15, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, R.; Lan, J.; Lin, S.; Li, P.; Gao, J.; Wang, Y.; Xie, Z.J.; Li, F.C.; Jiang, S.J. IGF2BP1 Significantly Enhances Translation Efficiency of Duck Hepatitis A Virus Type 1 without Affecting Viral Replication. Biomolecules 2019, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Rong, L.; Lu, J.; Pan, Q.; Liang, C. Insulin-like growth factor II mRNA binding protein 1 associates with Gag protein of human immunodeficiency virus type 1, and its overexpression affects virus assembly. J. Virol. 2008, 82, 5683–5692. [Google Scholar] [CrossRef]
- Zhou, Y.; Rong, L.; Zhang, J.; Aloysius, C.; Pan, Q.; Liang, C. Insulin-like growth factor II mRNA binding protein 1 modulates Rev-dependent human immunodeficiency virus type 1 RNA expression. Virology 2009, 393, 210–220. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, P.; Mao, K.; He, C.; Xu, Q.; Zhang, M.; Liu, H.; Zhou, Z.; Zhou, Q.; Zhou, Q.; et al. Anti-oncogene PTPN13 inactivation by hepatitis B virus X protein counteracts IGF2BP1 to promote hepatocellular carcinoma progression. Oncogene 2021, 40, 28–45. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center &Research Institute at Christiana Care Health Services; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y. LiquidAssociation: LiquidAssociation, R Package Version 1.53.0. 2022. Available online: https://bioconductor.org/packages/release/bioc/html/LiquidAssociation.html (accessed on 21 June 2023).
- Therneau, T.M. A Package for Survival Analysis in R, R package version 3.5-5. 2023. Available online: https://cran.r-project.org/web/packages/survival/vignettes/survival.pdf (accessed on 21 June 2023).
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Ding, J.; Qi, J.; Gu, Z. Characteristics and prognostic value of potential dependency genes in clear cell renal cell carcinoma based on a large-scale CRISPR-Cas9 and RNAi screening database DepMap. Int. J. Med. Sci. 2021, 18, 2063–2075. [Google Scholar] [CrossRef]
- Morgan, M.; Shepherd, L. ExperimentHub: Client to Access Experimenthub Resources, R Package Version 2.7.1. 2023. Available online: https://bioconductor.org/packages/release/bioc/html/ExperimentHub.html (accessed on 21 June 2023).
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45, D635–D642. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef] [PubMed]
- Megahed, F.A.K.; Zhou, X.; Sun, P. The Interactions between HBV and the Innate Immunity of Hepatocytes. Viruses 2020, 12, 285. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Li, W.; Miao, X.; Qi, Z.; Zeng, W.; Liang, J.; Liang, Z. Hepatitis B virus X protein upregulates HSP90alpha expression via activation of c-Myc in human hepatocarcinoma cell line, HepG2. Virol. J. 2010, 7, 45. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, Y.; Toh, S.T.; Sung, W.K.; Tan, P.; Chow, P.; Chung, A.Y.; Jooi, L.L.; Lee, C.G. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3. J. Hepatol. 2010, 53, 57–66. [Google Scholar] [CrossRef]
- You, X.; Liu, F.; Zhang, T.; Lv, N.; Liu, Q.; Shan, C.; Du, Y.; Kong, G.; Wang, T.; Ye, L.; et al. Hepatitis B virus X protein upregulates Lin28A/Lin28B through Sp-1/c-Myc to enhance the proliferation of hepatoma cells. Oncogene 2014, 33, 449–460. [Google Scholar] [CrossRef]
- Chang, T.C.; Zeitels, L.R.; Hwang, H.W.; Chivukula, R.R.; Wentzel, E.A.; Dews, M.; Jung, J.; Gao, P.; Dang, C.V.; Beer, M.A.; et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 3384–3389. [Google Scholar] [CrossRef] [PubMed]
- Li, K.C. Genome-wide coexpression dynamics: Theory and application. Proc. Natl. Acad. Sci. USA 2002, 99, 16875–16880. [Google Scholar] [CrossRef]
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018, 29, 25–38. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology 2002, 36, 74–83. [Google Scholar]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Barth, H.; Schuster, C.; Baumert, T.F. Hepatitis C virus entry: Molecular mechanisms and targets for antiviral therapy. Front. Biosci. (Landmark Ed.) 2009, 14, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [CrossRef]
- Kieft, J.S.; Zhou, K.; Jubin, R.; Doudna, J.A. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 2001, 7, 194–206. [Google Scholar] [CrossRef]
- Cheng, M.; Si, Y.; Niu, Y.; Liu, X.; Li, X.; Zhao, J.; Jin, Q.; Yang, W. High-throughput profiling of alpha interferon- and interleukin-28B-regulated microRNAs and identification of let-7s with anti-hepatitis C virus activity by targeting IGF2BP1. J. Virol. 2013, 87, 9707–9718. [Google Scholar] [CrossRef]
- Boyerinas, B.; Park, S.M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008, 68, 2587–2591. [Google Scholar] [CrossRef]
- Li, Y.; Masaki, T.; Shimakami, T.; Lemon, S.M. hnRNP L and NF90 interact with hepatitis C virus 5’-terminal untranslated RNA and promote efficient replication. J. Virol. 2014, 88, 7199–7209. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 2010, 84, 6615–6625. [Google Scholar] [CrossRef]
- Bradrick, S.S.; Nagyal, S.; Novatt, H. A miRNA-responsive cell-free translation system facilitates isolation of hepatitis C virus miRNP complexes. RNA 2013, 19, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Croonenborghs, T.; Roca Suarez, A.A.; Van Renne, N.; Jühling, F.; Oudot, M.A.; Virzì, A.; Bandiera, S.; Jamey, C.; Meszaros, G.; et al. Combined Analysis of Metabolomes, Proteomes, and Transcriptomes of Hepatitis C Virus-Infected Cells and Liver to Identify Pathways Associated With Disease Development. Gastroenterology 2019, 157, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. S5), 55–70. [Google Scholar] [CrossRef] [PubMed]
- van Boerdonk, R.A.; Daniels, J.M.; Bloemena, E.; Krijgsman, O.; Steenbergen, R.D.; Brakenhoff, R.H.; Grünberg, K.; Ylstra, B.; Meijer, C.J.; Smit, E.F.; et al. High-risk human papillomavirus-positive lung cancer: Molecular evidence for a pattern of pulmonary metastasis. J. Thorac. Oncol. 2013, 8, 711–718. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Ando, M.; Kubo, A.; Isa, S.; Yamamoto, S.; Tsujino, K.; Kurata, T.; Ou, S.H.; Takada, M.; Kawaguchi, T. Human papilloma virus in non-small cell lung cancer in never smokers: A systematic review of the literature. Lung Cancer 2014, 83, 8–13. [Google Scholar] [CrossRef]
- Khodabandehlou, N.; Mostafaei, S.; Etemadi, A.; Ghasemi, A.; Payandeh, M.; Hadifar, S.; Norooznezhad, A.H.; Kazemnejad, A.; Moghoofei, M. Human papilloma virus and breast cancer: The role of inflammation and viral expressed proteins. BMC Cancer 2019, 19, 61. [Google Scholar] [CrossRef]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef]
- Berman, T.A.; Schiller, J.T. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer 2017, 123, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Perrone, F.; Bossi, P.; Suardi, S.; Mariani, L.; Artusi, R.; Oggionni, M.; Rossini, C.; Cantù, G.; Squadrelli, M.; et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J. Clin. Oncol. 2006, 24, 5630–5636. [Google Scholar] [CrossRef]
- Li, W.; Thompson, C.H.; O’Brien, C.J.; McNeil, E.B.; Scolyer, R.A.; Cossart, Y.E.; Veness, M.J.; Walker, D.M.; Morgan, G.J.; Rose, B.R. Human papillomavirus positivity predicts favourable outcome for squamous carcinoma of the tonsil. Int. J. Cancer 2003, 106, 553–558. [Google Scholar] [CrossRef]
- Mellin, H.; Dahlgren, L.; Munck-Wikland, E.; Lindholm, J.; Rabbani, H.; Kalantari, M.; Dalianis, T. Human papillomavirus type 16 is episomal and a high viral load may be correlated to better prognosis in tonsillar cancer. Int. J. Cancer 2002, 102, 152–158. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.G.; Lee, D.; Kim, J.; Seo, T.; Choe, J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J. Biol. Chem. 2002, 277, 2923–2930. [Google Scholar] [CrossRef]
- Hussen, B.M.; Ahmadi, G.; Marzban, H.; Fard Azar, M.E.; Sorayyayi, S.; Karampour, R.; Nahand, J.S.; Hidayat, H.J.; Moghoofei, M. The role of HPV gene expression and selected cellular MiRNAs in lung cancer development. Microb. Pathog. 2021, 150, 104692. [Google Scholar] [CrossRef]
- Laban, S.; Gangkofner, D.S.; Holzinger, D.; Schroeder, L.; Eichmüller, S.B.; Zörnig, I.; Jäger, D.; Wichmann, G.; Dietz, A.; Broglie, M.A.; et al. Antibody Responses to Cancer Antigens Identify Patients with a Poor Prognosis among HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma Patients. Clin. Cancer Res. 2019, 25, 7405–7412. [Google Scholar] [CrossRef]
- Yu, L.; Lobanov, A.; Zheng, Z.M. Reply to Wang et al., Assessment of the Abundance and Potential Function of Human Papillomavirus Type 16 Circular E7 RNA. mBio 2022, 13, e0075822. [Google Scholar] [CrossRef]
- Murakami, T.; Ono, A. HIV-1 entry: Duels between Env and host antiviral transmembrane proteins on the surface of virus particles. Curr. Opin. Virol. 2021, 50, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T. Roles of the interactions between Env and Gag proteins in the HIV-1 replication cycle. Microbiol. Immunol. 2008, 52, 287–295. [Google Scholar] [CrossRef]
- Milev, M.P.; Brown, C.M.; Mouland, A.J. Live cell visualization of the interactions between HIV-1 Gag and the cellular RNA-binding protein Staufen1. Retrovirology 2010, 7, 41. [Google Scholar] [CrossRef]
- Rausch, J.W.; Le Grice, S.F. HIV Rev Assembly on the Rev Response Element (RRE): A Structural Perspective. Viruses 2015, 7, 3053–3075. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed]
- Huseynov, A.; Akin, I.; Duerschmied, D.; Scharf, R.E. Cardiac Arrhythmias in Post-COVID Syndrome: Prevalence, Pathology, Diagnosis, and Treatment. Viruses 2023, 15, 389. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Azevedo, R.B.; Botelho, B.G.; Hollanda, J.V.G.; Ferreira, L.V.L.; Junqueira de Andrade, L.Z.; Oei, S.S.M.L.; Mello, T.S.; Muxfeldt, E.S. COVID-19 and the cardiovascular system: A comprehensive review. J. Hum. Hypertens. 2021, 35, 4–11. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Gu, L.; Ma, L.; Duan, Y. Atlas of ACE2 gene expression reveals novel insights into transmission of SARS-CoV-2. Heliyon 2021, 7, e05850. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Hernandez, K.; Huang, L.; Luke, J.J. ACE2 and TMPRSS2 expression by clinical, HLA, immune, and microbial correlates across 34 human cancers and matched normal tissues: Implications for SARS-CoV-2 COVID-19. J. Immunother. Cancer 2020, 8, e001020. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyan, B.; Larabee, J.L.; Bodas, M.; Moore, A.R.; Burgett, A.W.G.; Myers, D.A.; Georgescu, C.; Wren, J.D.; Papin, J.F.; Walters, M.S. Characterization of the SARS-CoV-2 Host Response in Primary Human Airway Epithelial Cells from Aged Individuals. Viruses 2021, 13, 1603. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Pietzsch, C.; Ramanathan, P.; Santos, R.I.; Ilinykh, P.A.; Garcia-Blanco, M.A.; Bukreyev, A.; Bradrick, S.S. Staufen1 Interacts with Multiple Components of the Ebola Virus Ribonucleoprotein and Enhances Viral RNA Synthesis. mBio 2018, 9, e01771-18. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Kwon, Y.K.; Joh, S.J.; Kwon, J.H.; Lindberg, A.M. Differential diagnosis between type-specific duck hepatitis virus type 1 (DHV-1) and recent Korean DHV-1-like isolates using a multiplex polymerase chain reaction. Avian Pathol. 2008, 37, 171–177. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Zhao, F.; Zhang, Q.; Zuo, W.; Guo, M.; Zhang, X.; Wu, Y. Identification and Functional Analyses of Host Proteins Interacting with the p17 Protein of Avian Reovirus. Viruses 2022, 14, 892. [Google Scholar] [CrossRef]
- Couteaudier, M.; Denesvre, C. Marek’s disease virus and skin interactions. Vet. Res. 2014, 45, 36. [Google Scholar] [CrossRef]
- Mitra, A.; Luo, J.; Zhang, H.; Cui, K.; Zhao, K.; Song, J. Marek’s disease virus infection induces widespread differential chromatin marks in inbred chicken lines. BMC Genom. 2012, 13, 557. [Google Scholar] [CrossRef]
- Mai, Y.; Gao, G. Expression of IMP1 enhances production of murine leukemia virus vector by facilitating viral genomic RNA packaging. PLoS ONE 2010, 5, e15881. [Google Scholar] [CrossRef]
- Jefferson, M.; Donaszi-Ivanov, A.; Pollen, S.; Dalmay, T.; Saalbach, G.; Powell, P.P. Host factors that interact with the pestivirus N-terminal protease, Npro, are components of the ribonucleoprotein complex. J. Virol. 2014, 88, 10340–10353. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, L.; Andruska, N.; Mao, C.; Le, J.; Shapiro, D.J. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl. Oncol. 2017, 10, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, Q.; Yang, H.; Zhang, X.W.; Feng, N.; Wang, J.K.; Liu, T.T.; Zeng, K.W.; Tu, P.F. Allosteric Regulation of IGF2BP1 as a Novel Strategy for the Activation of Tumor Immune Microenvironment. ACS Cent. Sci. 2022, 8, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, J.B.; Müller, S.; Hüttelmaier, S. RNA-binding proteins in cancer drug discovery. Drug Discov. Today 2023, 28, 103580. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Impact of IGF2BP1 on Virus | Impact of Virus on IGF2BP1 |
|---|---|---|
| HBV | Binding to c-Myc mRNA ↑ [49] | |
| RNA/protein expression ↑ [49] | ||
| HCV | Translation ↑ [45] | Protein expression ↑ [45] |
| Replication ↑ [86] | ||
| HPV | Stability of E7 RNA ↑ [43] | Heat stability ↓ [43] |
| HIV-1 | Virus production↓ [47,48] | |
| Infectivity ↓ [47,48] | ||
| SARS-CoV-2 | RNA levels ↑ [44] | RNA expression ↑ [44] |
| Stability of S RNA ↑ [44] | ||
| ZIKV | RNA levels ↑ [44] | |
| EBOV | Infectivity ↑ [120] | |
| DHV-1 | Translation ↑ [46] | Protein expression ↑ [46] |
| ARV | Replication ↑ [122] | RNA expression ↑ [122] |
| MDV | RNA expression ↓ [124] | |
| MLV | stability of genomic RNA ↑ [125] | |
| Protein levels ↓ [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glaß, M.; Hüttelmaier, S. IGF2BP1—An Oncofetal RNA-Binding Protein Fuels Tumor Virus Propagation. Viruses 2023, 15, 1431. https://doi.org/10.3390/v15071431
Glaß M, Hüttelmaier S. IGF2BP1—An Oncofetal RNA-Binding Protein Fuels Tumor Virus Propagation. Viruses. 2023; 15(7):1431. https://doi.org/10.3390/v15071431
Chicago/Turabian StyleGlaß, Markus, and Stefan Hüttelmaier. 2023. "IGF2BP1—An Oncofetal RNA-Binding Protein Fuels Tumor Virus Propagation" Viruses 15, no. 7: 1431. https://doi.org/10.3390/v15071431
APA StyleGlaß, M., & Hüttelmaier, S. (2023). IGF2BP1—An Oncofetal RNA-Binding Protein Fuels Tumor Virus Propagation. Viruses, 15(7), 1431. https://doi.org/10.3390/v15071431