
Anionic Pulmonary Surfactant Lipid Treatment Inhibits Rhinovirus A Infection of the Human Airway Epithelium

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture and Virus
2.2. Pulmonary Surfactant Lipid Treatment and Viral Infection of Differentiated AEC Cultures
2.3. Quantification of RV-A16 Genomic Copies and Host Gene Expression using qRT-PCR
2.4. Whole-Transcriptome RNA Sequencing and Differential Expression Analysis
2.5. Pathway and Cell-Type Enrichment Analyses
2.6. Immunofluorescent Staining
2.7. Evaluation of Virus Internalization in AECs using the Trypsinization Method
2.8. Statistical Analysis
3. Results
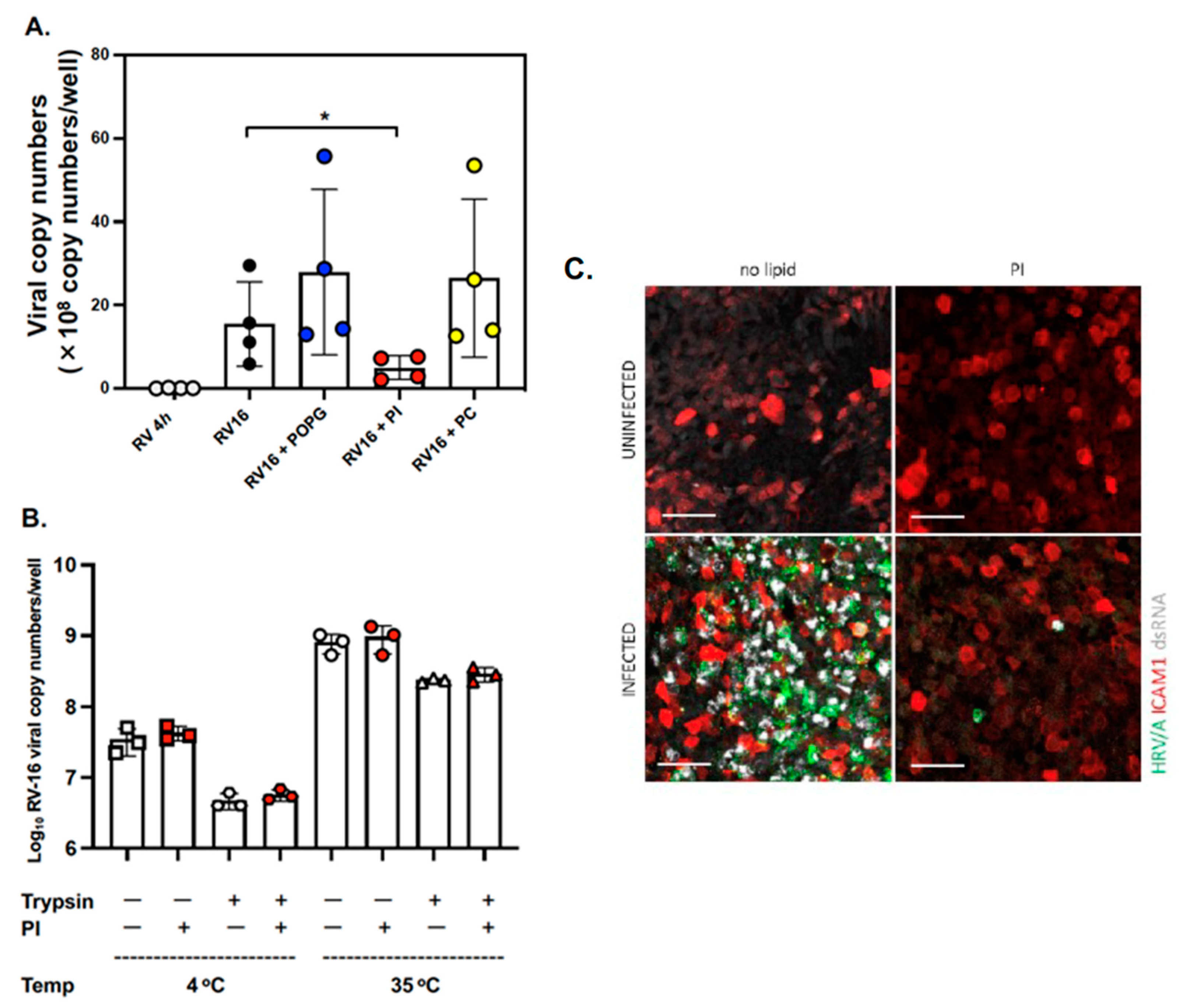
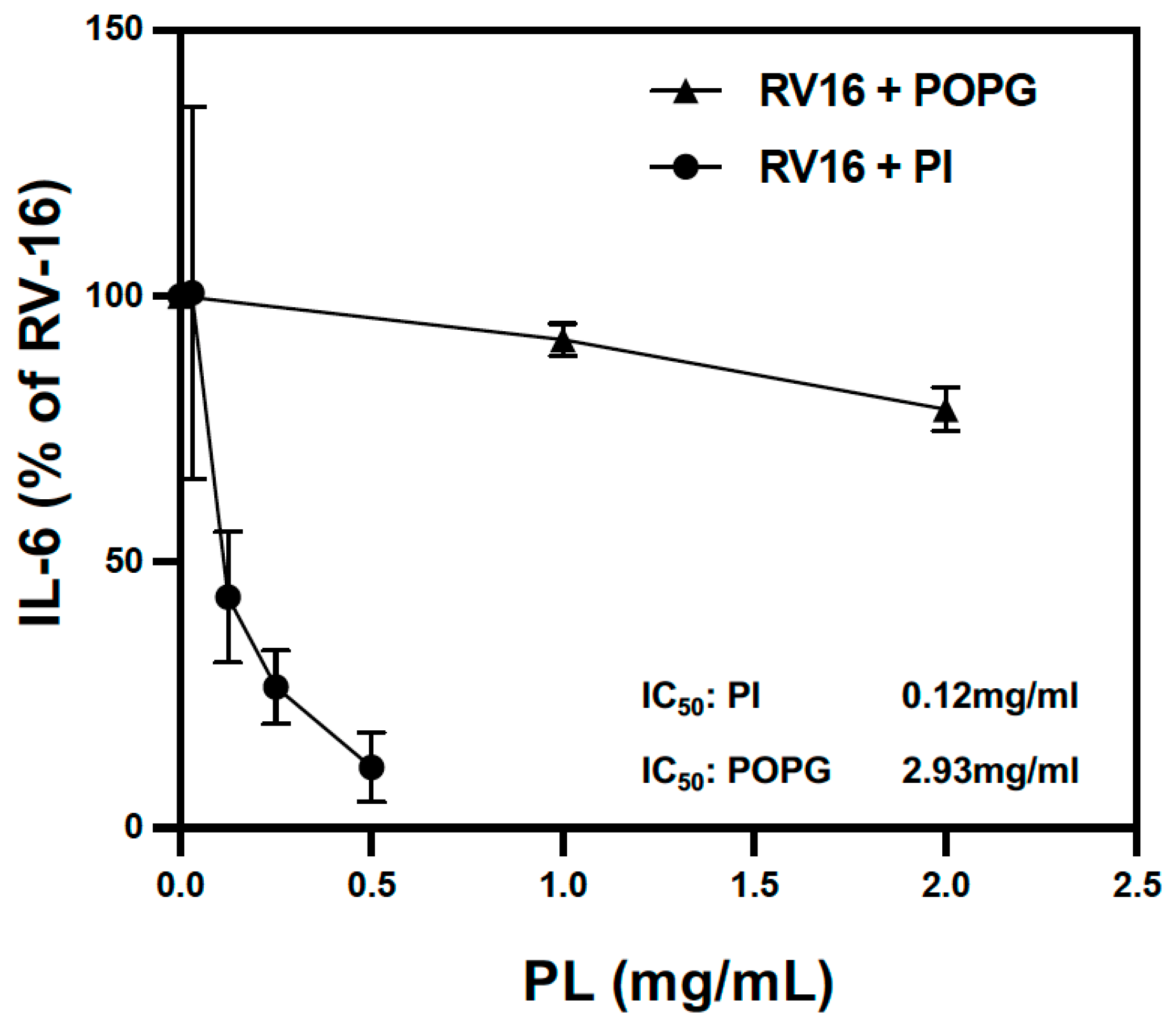
3.1. The Anionic Surfactant Lipid, PI, Inhibits RV-A16 Replication in Human AEC Cultures
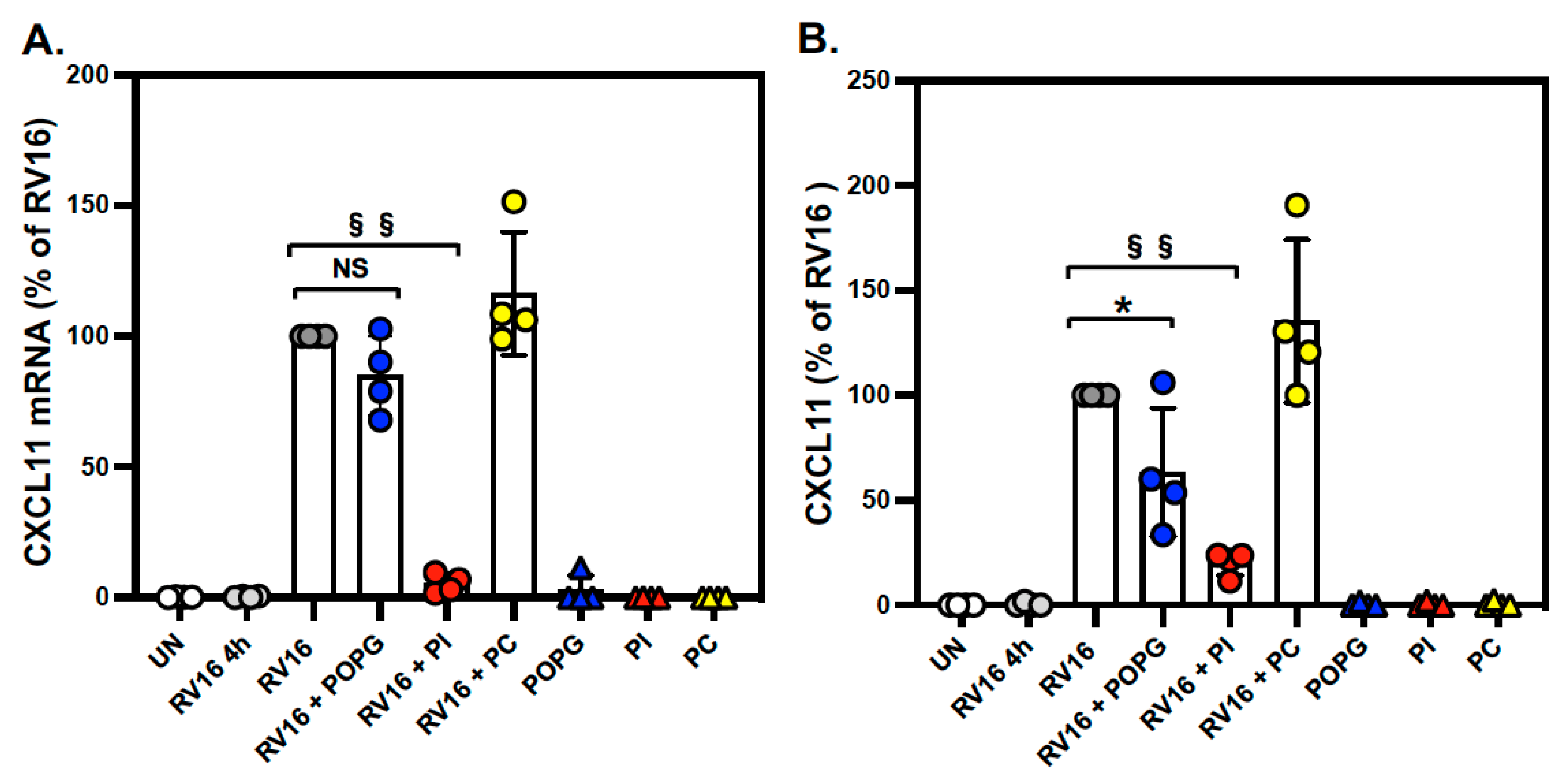
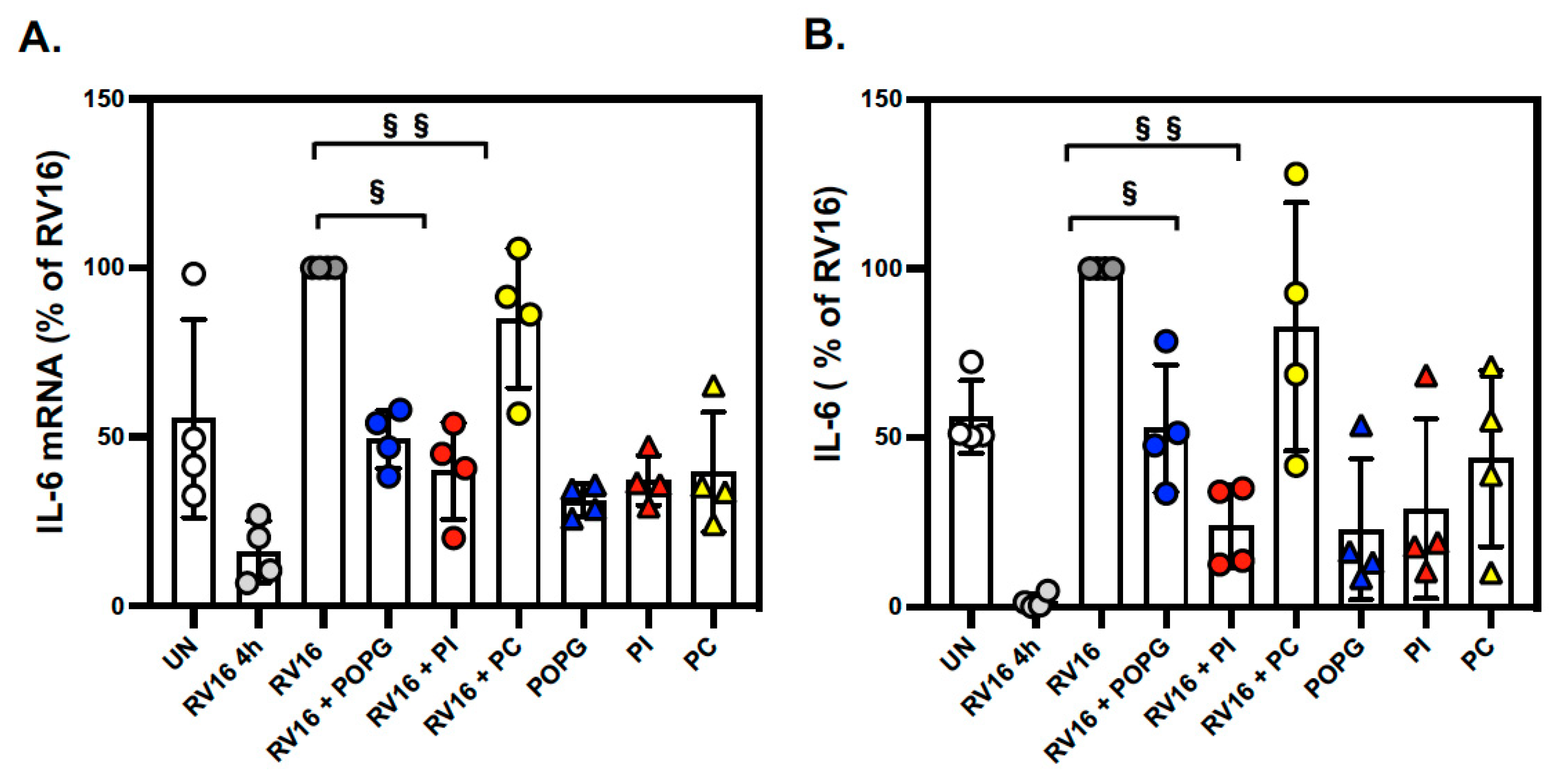
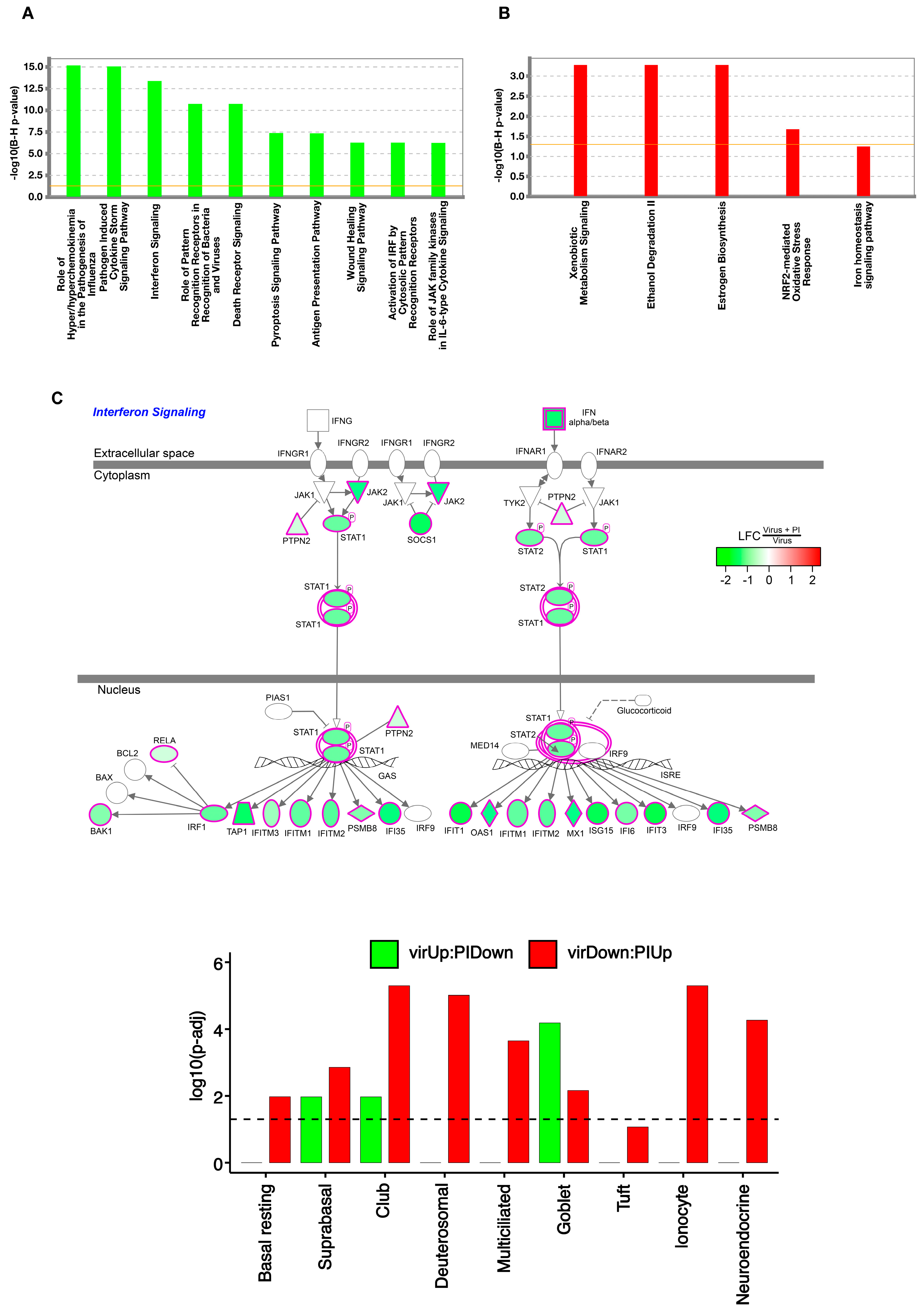
3.2. Pulmonary Surfactant Lipids Reduce Antiviral and Proinflammatory Responses in Human AEC Cultures
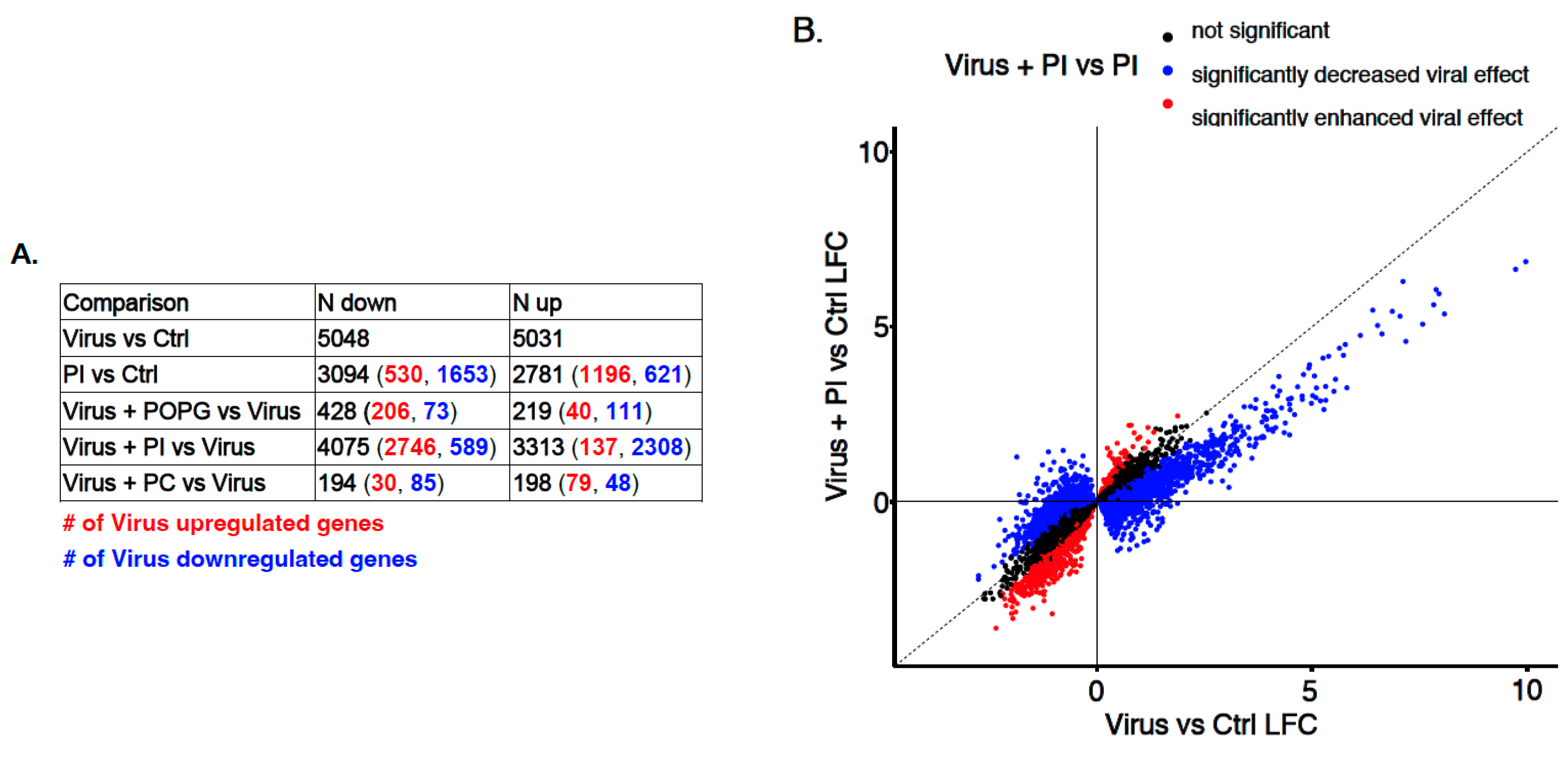
3.3. Transcriptome-Wide Epithelial Responses to RV-A16 Infection Attenuated by PI Pretreatment
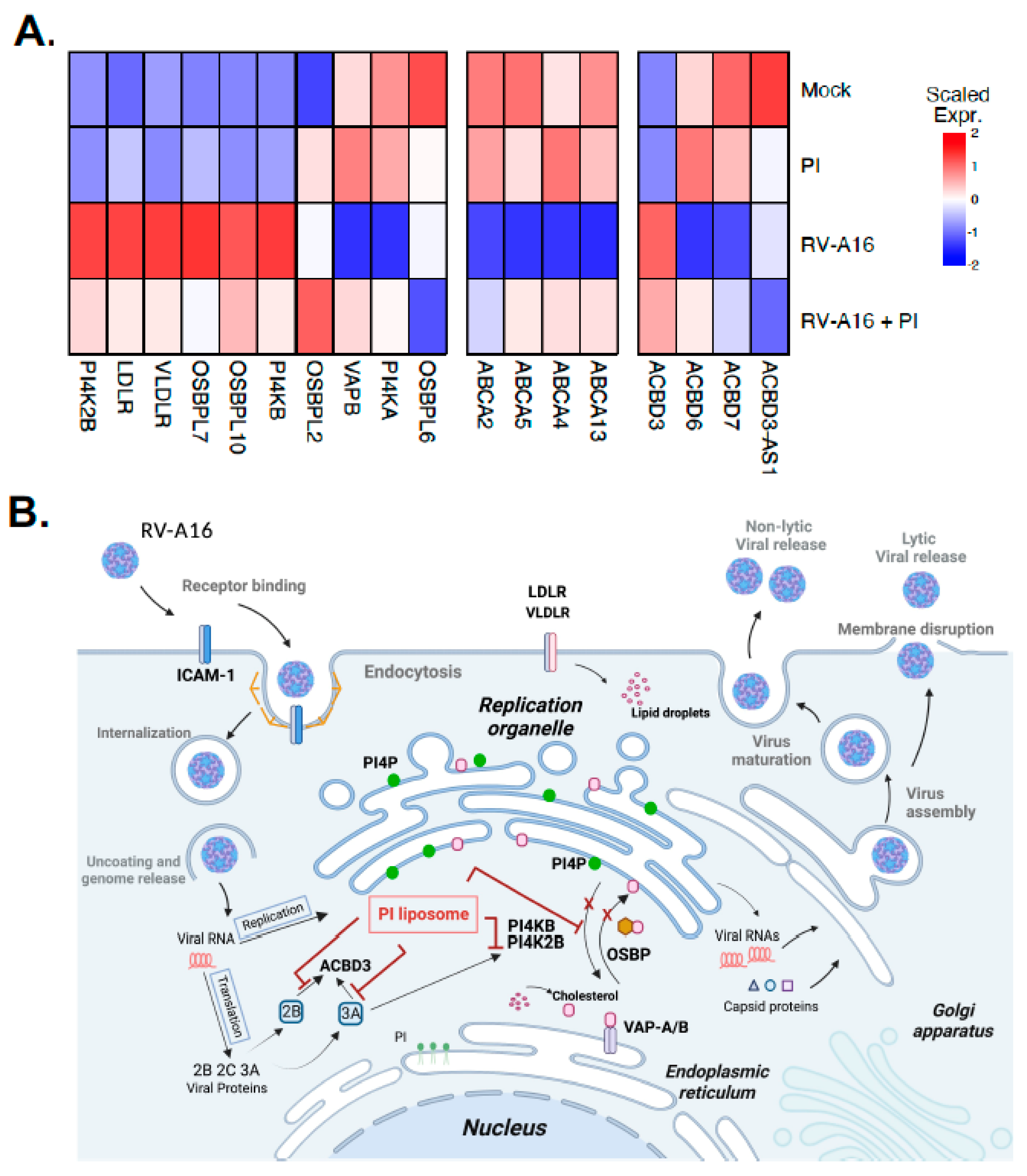
3.4. PI Inhibits RV-A16 Mediated Regulation of Genes Critical for the Viral Replication Organelle (RO) Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCA gene | ATP-binding cassette transporter gene |
| ACBD3 | Acyl CoA-binding domain 3 |
| ALI | air–liquid interface culture |
| RV-16 | human rhinovirus A16 |
| CXCL11 | C-X-C motif chemokine 11 |
| DGE | differential gene expression |
| DPPC | dipalmitoyl-phosphatidylcholine |
| TGN | Golgi and trans-Golgi network |
| pH1N1-IAV | H1N1 A/California/07/2009 |
| H3N2-IAV | H3N2-IAV (Philippines 82/H3N2) |
| PI4K | human phosphatidylinositol 4-kinases |
| PI4K2A | human phosphatidylinositol 4-kinases type 2 alpha |
| PI4K2B | human phosphatidylinositol 4-kinases type 2 beta |
| PI4KA | human phosphatidylinositol 4-kinase IIIα |
| PI4KB | phosphatidylinositol 4-kinase IIIβ |
| IL-6 | interleukin-6 |
| IFITM1 | interferon-induced transmembrane protein 1 |
| IFITM2 | interferon-induced transmembrane protein 2 |
| IFITM3 | interferon-induced transmembrane protein 3 |
| IRF1 | interferon regulatory factor 1 |
| IRF7 | interferon regulatory factor 7 |
| moi | multiplicity of infection |
| OAS1 | 2′-5′-oligoadenylate synthetase 1 |
| OSBP | oxysterol-binding protein |
| POPG | palmitoyl-oleoyl-phosphatidylglycerol |
| POPC | palmitoyl-oleoyl-phosphatidylcholine |
| AECs | primary human trachea cells |
| RO | replication organelle |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SOCS1 | suppressor of cytokine signaling 1 |
| STAT1 | signal transducer and activator of transcription 1 |
| STAT2 | signal transducer and activator of transcription 2 |
| COVID-19 | the coronavirus disease 2019 |
| TLRs | toll-like receptors |
| VAP-A | vesicle-associated membrane protein-associated protein isoforms A |
| VAPVAP-B | vesicle-associated membrane protein-associated protein isoforms B |
| VLDLR | very-low-density lipoprotein receptor |
References
- Friedlander, S.L.; Busse, W.W. The role of rhinovirus in asthma exacerbations. J. Allergy Clin. Immunol. 2005, 116, 267–273. [Google Scholar] [CrossRef]
- Basnet, S.; Palmenberg, A.C.; Gern, J.E. Rhinoviruses and Their Receptors. Chest 2019, 155, 1018–1025. [Google Scholar] [CrossRef]
- Wark, P.A.B. Asthma and the Dysregulated Immune Response to Rhinovirus. Am. J. Respir. Crit. Care Med. 2020, 202, 157–159. [Google Scholar] [CrossRef]
- Lee, W.M.; Lemanske, R.F., Jr.; Evans, M.D.; Vang, F.; Pappas, T.; Gangnon, R.; Jackson, D.J.; Gern, J.E. Human rhinovirus species and season of infection determine illness severity. Am. J. Respir. Crit. Care Med. 2012, 186, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Lamborn, I.T.; Jing, H.; Zhang, Y.; Drutman, S.B.; Abbott, J.K.; Munir, S.; Bade, S.; Murdock, H.M.; Santos, C.P.; Brock, L.G.; et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 2017, 214, 1949–1972. [Google Scholar] [CrossRef] [PubMed]
- Iwane, M.K.; Prill, M.M.; Lu, X.; Miller, E.K.; Edwards, K.M.; Hall, C.B.; Griffin, M.R.; Staat, M.A.; Anderson, L.J.; Williams, J.V.; et al. Human rhinovirus species associated with hospitalizations for acute respiratory illness in young US children. J. Infect. Dis. 2011, 204, 1702–1710. [Google Scholar] [CrossRef]
- Cox, D.W.; Bizzintino, J.; Ferrari, G.; Khoo, S.K.; Zhang, G.; Whelan, S.; Lee, W.M.; Bochkov, Y.A.; Geelhoed, G.C.; Goldblatt, J.; et al. Human rhinovirus species C infection in young children with acute wheeze is associated with increased acute respiratory hospital admissions. Am. J. Respir. Crit. Care Med. 2013, 188, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef]
- Kennedy, J.L.; Pham, S.; Borish, L. Rhinovirus and Asthma Exacerbations. Immunol. Allergy Clin. N. Am. 2019, 39, 335–344. [Google Scholar] [CrossRef]
- Bergroth, E.; Aakula, M.; Elenius, V.; Remes, S.; Piippo-Savolainen, E.; Korppi, M.; Piedra, P.A.; Bochkov, Y.A.; Gern, J.E.; Camargo, C.A., Jr.; et al. Rhinovirus Type in Severe Bronchiolitis and the Development of Asthma. J. Allergy Clin. Immunol. Pract. 2020, 8, 588–595. [Google Scholar] [CrossRef]
- Jartti, T.; Gern, J.E. Rhinovirus-associated wheeze during infancy and asthma development. Curr. Respir. Med. Rev. 2011, 7, 160–166. [Google Scholar] [CrossRef]
- Cox, D.W.; Khoo, S.K.; Zhang, G.; Lindsay, K.; Keil, A.D.; Knight, G.; Gern, J.E.; Laing, I.A.; Bizzintino, J.; Le Souef, P.N. Rhinovirus is the most common virus and rhinovirus-C is the most common species in paediatric intensive care respiratory admissions. Eur. Respir. J. 2018, 52, 1800207. [Google Scholar] [CrossRef] [PubMed]
- Raita, Y.; Camargo, C.A., Jr.; Bochkov, Y.A.; Celedon, J.C.; Gern, J.E.; Mansbach, J.M.; Rhee, E.P.; Freishtat, R.J.; Hasegawa, K. Integrated-omics endotyping of infants with rhinovirus bronchiolitis and risk of childhood asthma. J. Allergy Clin. Immunol. 2021, 147, 2108–2117. [Google Scholar] [CrossRef]
- Numata, M.; Voelker, D.R. Anti-inflammatory and anti-viral actions of anionic pulmonary surfactant phospholipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159139. [Google Scholar] [CrossRef] [PubMed]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef]
- Hashimoto, M.; Asai, Y.; Ogawa, T. Treponemal phospholipids inhibit innate immune responses induced by pathogen-associated molecular patterns. J. Biol. Chem. 2003, 278, 44205–44213. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, P.; Numata, M.; Berry, K.Z.; Fickes, R.; Leslie, C.C.; Murphy, R.C.; Voelker, D.R. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling. J. Lipid Res. 2016, 57, 993–1005. [Google Scholar] [CrossRef]
- Numata, M.; Chu, H.W.; Dakhama, A.; Voelker, D.R. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc. Natl. Acad. Sci. USA 2010, 107, 320–325. [Google Scholar] [CrossRef]
- Numata, M.; Kandasamy, P.; Nagashima, Y.; Fickes, R.; Murphy, R.C.; Voelker, D.R. Phosphatidylinositol inhibits respiratory syncytial virus infection. J. Lipid Res. 2015, 56, 578–587. [Google Scholar] [CrossRef]
- Numata, M.; Nagashima, Y.; Moore, M.L.; Berry, K.Z.; Chan, M.; Kandasamy, P.; Peebles, R.S., Jr.; Murphy, R.C.; Voelker, D.R. Phosphatidylglycerol provides short-term prophylaxis against respiratory syncytial virus infection. J. Lipid Res. 2013, 54, 2133–2143. [Google Scholar] [CrossRef]
- Numata, M.; Mitchell, J.R.; Tipper, J.L.; Brand, J.D.; Trombley, J.E.; Nagashima, Y.; Kandasamy, P.; Chu, H.W.; Harrod, K.S.; Voelker, D.R. Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models. J. Biol. Chem. 2020, 295, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Everman, J.L.; Sajuthi, S.; Saef, B.; Rios, C.; Stoner, A.M.; Numata, M.; Hu, D.; Eng, C.; Oh, S.; Rodriguez-Santana, J.; et al. Functional genomics of CDHR3 confirms its role in HRV-C infection and childhood asthma exacerbations. J. Allergy Clin. Immunol. 2019, 144, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Wang, W.; Rueckert, R.R. Complete sequence of the RNA genome of human rhinovirus 16, a clinically useful common cold virus belonging to the ICAM-1 receptor group. Virus Genes 1995, 9, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Mosser, A.G.; Brockman-Schneider, R.; Amineva, S.; Burchell, L.; Sedgwick, J.B.; Busse, W.W.; Gern, J.E. Similar frequency of rhinovirus-infectible cells in upper and lower airway epithelium. J. Infect. Dis. 2002, 185, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Guedán, A.; Swieboda, D.; Charles, M.; Toussaint, M.; Johnston, S.L.; Asfor, A.; Panjwani, A.; Tuthill, T.J.; Danahay, H.; Raynham, T.; et al. Investigation of the Role of Protein Kinase D in Human Rhinovirus Replication. J. Virol. 2017, 91, e00217-17. [Google Scholar] [CrossRef]
- Numata, M.; Kandasamy, P.; Nagashima, Y.; Posey, J.; Hartshorn, K.; Woodland, D.; Voelker, D.R. Phosphatidylglycerol suppresses influenza A virus infection. Am. J. Respir. Cell Mol. Biol. 2012, 46, 479–487. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Hanson, K.M.; Keles, S.; Brockman-Schneider, R.A.; Jarjour, N.N.; Gern, J.E. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010, 3, 69–80. [Google Scholar] [CrossRef]
- Schaefer, N.; Li, X.; Seibold, M.A.; Jarjour, N.N.; Denlinger, L.C.; Castro, M.; Coverstone, A.M.; Teague, W.G.; Boomer, J.; Bleecker, E.R.; et al. The effect of BPIFA1/SPLUNC1 genetic variation on its expression and function in asthmatic airway epithelium. JCI Insight 2019, 4, e127237. [Google Scholar] [CrossRef]
- Sajuthi, S.P.; Everman, J.L.; Jackson, N.D.; Saef, B.; Rios, C.L.; Moore, C.M.; Mak, A.C.Y.; Eng, C.; Fairbanks-Mahnke, A.; Salazar, S.; et al. Nasal airway transcriptome-wide association study of asthma reveals genetically driven mucus pathobiology. Nat. Commun. 2022, 13, 1632. [Google Scholar] [CrossRef]
- Jackson, N.D.; Everman, J.L.; Chioccioli, M.; Feriani, L.; Goldfarbmuren, K.C.; Sajuthi, S.P.; Rios, C.L.; Powell, R.; Armstrong, M.; Gomez, J.; et al. Single-Cell and Population Transcriptomics Reveal Pan-epithelial Remodeling in Type 2-High Asthma. Cell Rep. 2020, 32, 107872. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sikkema, L.; Strobl, D.; Zappia, L.; Madissoon, E.; Markov, N.; Zaragosi, L.; Ansari, M.; Arguel, M.; Apperloo, L.; Bécavin, C.; et al. An integrated cell atlas of the human lung in health and disease. bioRxiv 2022. [Google Scholar] [CrossRef]
- Vladar, E.K.; Brody, S.L. Analysis of ciliogenesis in primary culture mouse tracheal epithelial cells. Methods Enzym. 2013, 525, 285–309. [Google Scholar] [CrossRef]
- Egli, A.; Santer, D.M.; O’Shea, D.; Tyrrell, D.L.; Houghton, M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg. Microbes Infect. 2014, 3, e51. [Google Scholar] [CrossRef] [PubMed]
- Sykes, A.; Macintyre, J.; Edwards, M.R.; Del Rosario, A.; Haas, J.; Gielen, V.; Kon, O.M.; McHale, M.; Johnston, S.L. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax 2014, 69, 240–246. [Google Scholar] [CrossRef]
- Khoo, S.K.; Read, J.; Franks, K.; Zhang, G.; Bizzintino, J.; Coleman, L.; McCrae, C.; Oberg, L.; Troy, N.M.; Prastanti, F.; et al. Upper Airway Cell Transcriptomics Identify a Major New Immunological Phenotype with Strong Clinical Correlates in Young Children with Acute Wheezing. J. Immunol. 2019, 202, 1845–1858. [Google Scholar] [CrossRef]
- Roulin, P.S.; Lotzerich, M.; Torta, F.; Tanner, L.B.; van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Strating, J.R.; van Kuppeveld, F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017, 47, 24–33. [Google Scholar] [CrossRef]
- van der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.; van Kuppeveld, F.J.M. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trends Microbiol. 2016, 24, 535–546. [Google Scholar] [CrossRef]
- McCrae, C.; Dzgoev, A.; Stahlman, M.; Horndahl, J.; Svard, R.; Grosse, A.; Grosskopf, T.; Skujat, M.A.; Williams, N.; Schubert, S.; et al. Lanosterol Synthase Regulates Human Rhinovirus Replication in Human Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2018, 59, 713–722. [Google Scholar] [CrossRef] [PubMed]
- McPhail, J.A.; Burke, J.E. Molecular mechanisms of PI4K regulation and their involvement in viral replication. Traffic 2022, 24, 131–145. [Google Scholar] [CrossRef]
- Essaidi-Laziosi, M.; Geiser, J.; Huang, S.; Constant, S.; Kaiser, L.; Tapparel, C. Interferon-Dependent and Respiratory Virus-Specific Interference in Dual Infections of Airway Epithelia. Sci. Rep. 2020, 10, 10246. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, W.; Ray, A.; Wu, Y.; Einarsson, O.; Landry, M.L.; Gwaltney, J., Jr.; Elias, J.A. Rhinovirus stimulation of interleukin-6 in vivo and in vitro. Evidence for nuclear factor kappa B-dependent transcriptional activation. J. Clin. Investig. 1996, 97, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, K.; Smits, H.H.; Timmers, M.C.; de Klerk, E.P.; Dolhain, R.J.; Dick, E.C.; Hiemstra, P.S.; Sterk, P.J. Experimental rhinovirus 16 infection. Effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am. J. Respir. Crit. Care Med. 1997, 156, 609–616. [Google Scholar] [CrossRef]
- Foxman, E.F.; Storer, J.A.; Vanaja, K.; Levchenko, A.; Iwasaki, A. Two interferon-independent double-stranded RNA-induced host defense strategies suppress the common cold virus at warm temperature. Proc. Natl. Acad. Sci. USA 2016, 113, 8496–8501. [Google Scholar] [CrossRef]
- Wang, Q.; Nagarkar, D.R.; Bowman, E.R.; Schneider, D.; Gosangi, B.; Lei, J.; Zhao, Y.; McHenry, C.L.; Burgens, R.V.; Miller, D.J. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 2009, 183, 6989–6997. [Google Scholar] [CrossRef] [PubMed]
- Quiner, C.A.; Jackson, W.T. Fragmentation of the Golgi apparatus provides replication membranes for human rhinovirus 1A. Virology 2010, 407, 185–195. [Google Scholar] [CrossRef]
- Glingston, R.S.; Deb, R.; Kumar, S.; Nagotu, S. Organelle dynamics and viral infections: At cross roads. Microbes Infect. 2019, 21, 20–32. [Google Scholar] [CrossRef]
- Gaudernak, E.; Seipelt, J.; Triendl, A.; Grassauer, A.; Kuechler, E. Antiviral effects of pyrrolidine dithiocarbamate on human rhinoviruses. J. Virol. 2002, 76, 6004–6015. [Google Scholar] [CrossRef]
- Tai, A.W.; Bojjireddy, N.; Balla, T. A homogeneous and nonisotopic assay for phosphatidylinositol 4-kinases. Anal. Biochem. 2011, 417, 97–102. [Google Scholar] [CrossRef]
- Ishikawa-Sasaki, K.; Nagashima, S.; Taniguchi, K.; Sasaki, J. Model of OSBP-Mediated Cholesterol Supply to Aichi Virus RNA Replication Sites Involving Protein-Protein Interactions among Viral Proteins, ACBD3, OSBP, VAP-A/B, and SAC1. J. Virol. 2018, 92, e01952-17. [Google Scholar] [CrossRef]
- Nchoutmboube, J.; Ford-Siltz, L.A.; Belov, G.A. Enterovirus replication: Go with the (counter) flow. Trends Microbiol. 2015, 23, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Lyoo, H.; van der Schaar, H.M.; Dorobantu, C.M.; Rabouw, H.H.; Strating, J.R.; Van Kuppeveld, F.J. ACBD3 is an essential pan-enterovirus host factor that mediates the interaction between viral 3A protein and cellular protein PI4KB. mBio 2019, 10, e02742-18. [Google Scholar] [CrossRef] [PubMed]
- Ilnytska, O.; Santiana, M.; Hsu, N.Y.; Du, W.L.; Chen, Y.H.; Viktorova, E.G.; Belov, G.; Brinker, A.; Storch, J.; Moore, C.; et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013, 14, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Roulin, P.S.; Murer, L.P.; Greber, U.F. A single point mutation in the rhinovirus 2B protein reduces the requirement for phosphatidylinositol 4-kinase class III beta in viral replication. J. Virol. 2018, 92, e01462-18. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Unger, S.D.; Zhang, N.; Taka, S.; Michel, S.; Akdag, N.; Lan, F.; Helfer, M.; Hudemann, C.; Eickmann, M.; et al. Development and characterization of DNAzyme candidates demonstrating significant efficiency against human rhinoviruses. J. Allergy Clin. Immunol. 2019, 143, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Phipps, K.M.; Martinez, A.; Lu, J.; Heinz, B.A.; Zhao, G. Small interfering RNA molecules as potential anti-human rhinovirus agents: In vitro potency, specificity, and mechanism. Antivir. Res. 2004, 61, 49–55. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numata, M.; Sajuthi, S.; Bochkov, Y.A.; Loeffler, J.; Everman, J.; Vladar, E.K.; Cooney, R.A.; Reinhardt, R.L.; Liu, A.H.; Seibold, M.A.; et al. Anionic Pulmonary Surfactant Lipid Treatment Inhibits Rhinovirus A Infection of the Human Airway Epithelium. Viruses 2023, 15, 747. https://doi.org/10.3390/v15030747
Numata M, Sajuthi S, Bochkov YA, Loeffler J, Everman J, Vladar EK, Cooney RA, Reinhardt RL, Liu AH, Seibold MA, et al. Anionic Pulmonary Surfactant Lipid Treatment Inhibits Rhinovirus A Infection of the Human Airway Epithelium. Viruses. 2023; 15(3):747. https://doi.org/10.3390/v15030747
Chicago/Turabian StyleNumata, Mari, Satria Sajuthi, Yury A. Bochkov, Jessica Loeffler, Jamie Everman, Eszter K. Vladar, Riley A. Cooney, Richard Lee Reinhardt, Andrew H. Liu, Max A. Seibold, and et al. 2023. "Anionic Pulmonary Surfactant Lipid Treatment Inhibits Rhinovirus A Infection of the Human Airway Epithelium" Viruses 15, no. 3: 747. https://doi.org/10.3390/v15030747
APA StyleNumata, M., Sajuthi, S., Bochkov, Y. A., Loeffler, J., Everman, J., Vladar, E. K., Cooney, R. A., Reinhardt, R. L., Liu, A. H., Seibold, M. A., & Voelker, D. R. (2023). Anionic Pulmonary Surfactant Lipid Treatment Inhibits Rhinovirus A Infection of the Human Airway Epithelium. Viruses, 15(3), 747. https://doi.org/10.3390/v15030747