


Putative Mitoviruses without In-Frame UGA(W) Codons: Evolutionary Implications


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Open Access Databases
2.2. Bioinformatic Tools
3. Results and Discussion
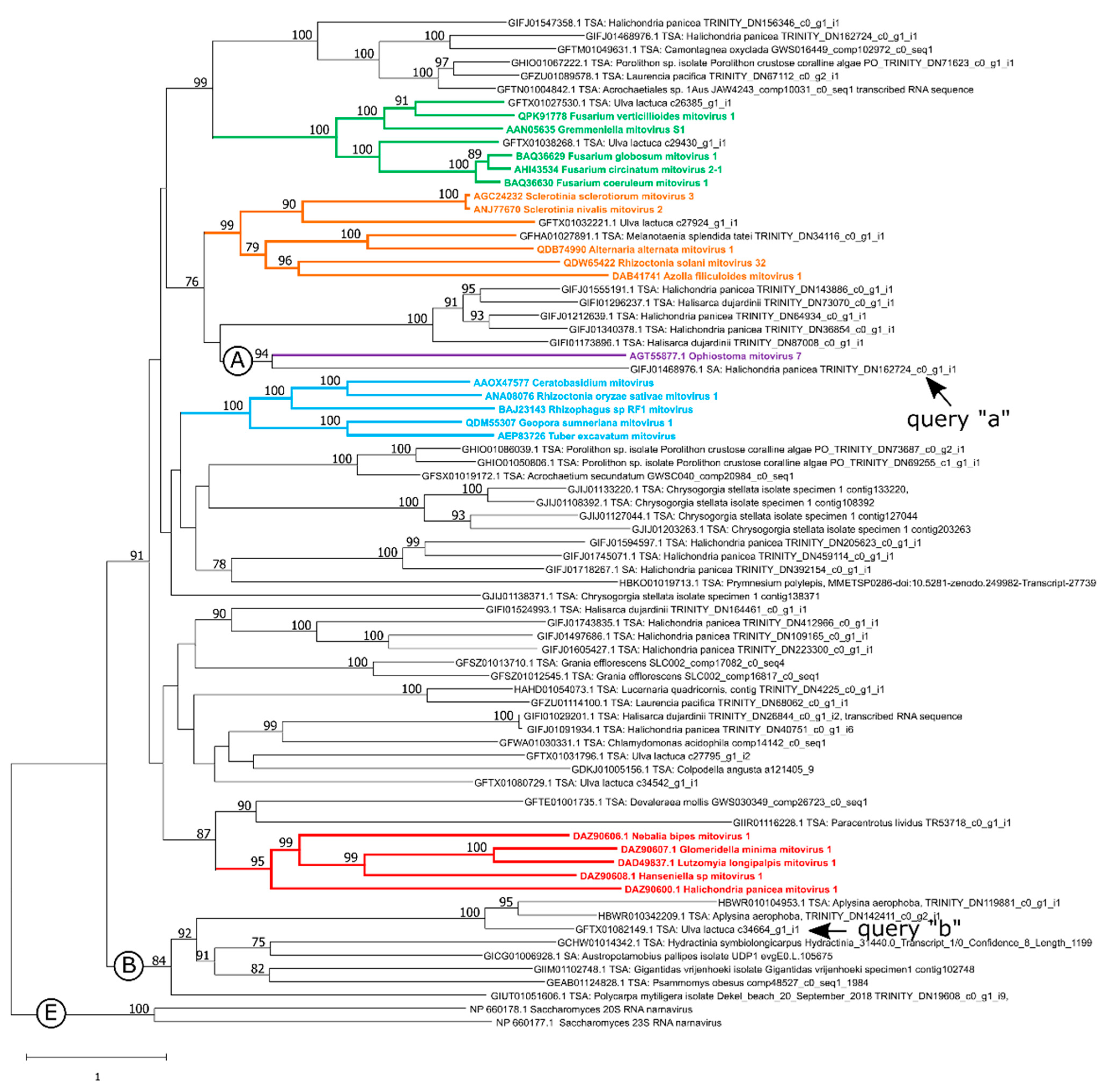
3.1. Search for Putative Mitoviruses in the Open Databases
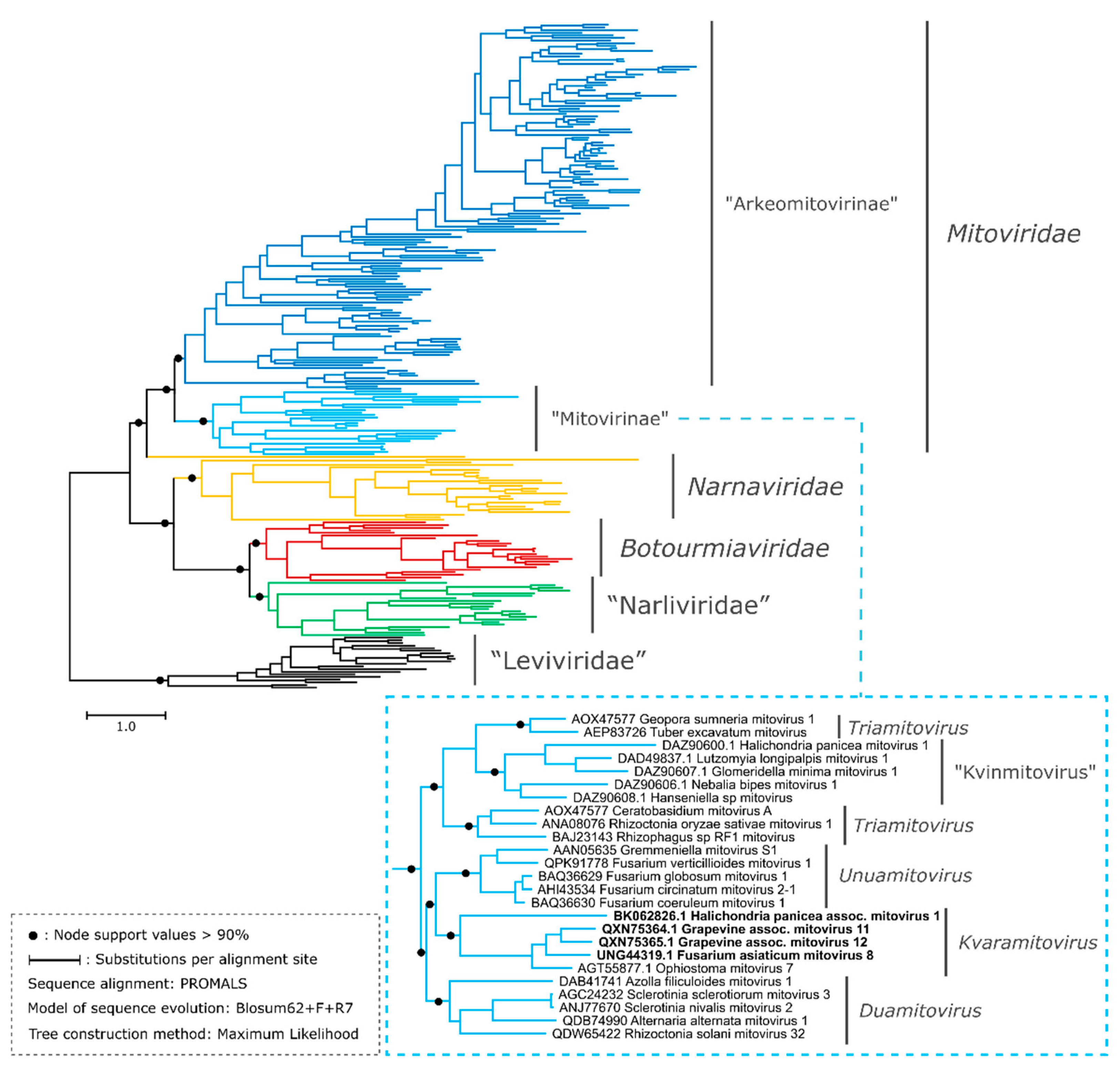
3.2. New Putative Mitoviruses in the Phylogenetic Context of the Phylum Lenarviricota
3.3. Proposal of Supra-Generic Taxa
3.4. The genus Kvaramitovirus: A Proposal for Expansion
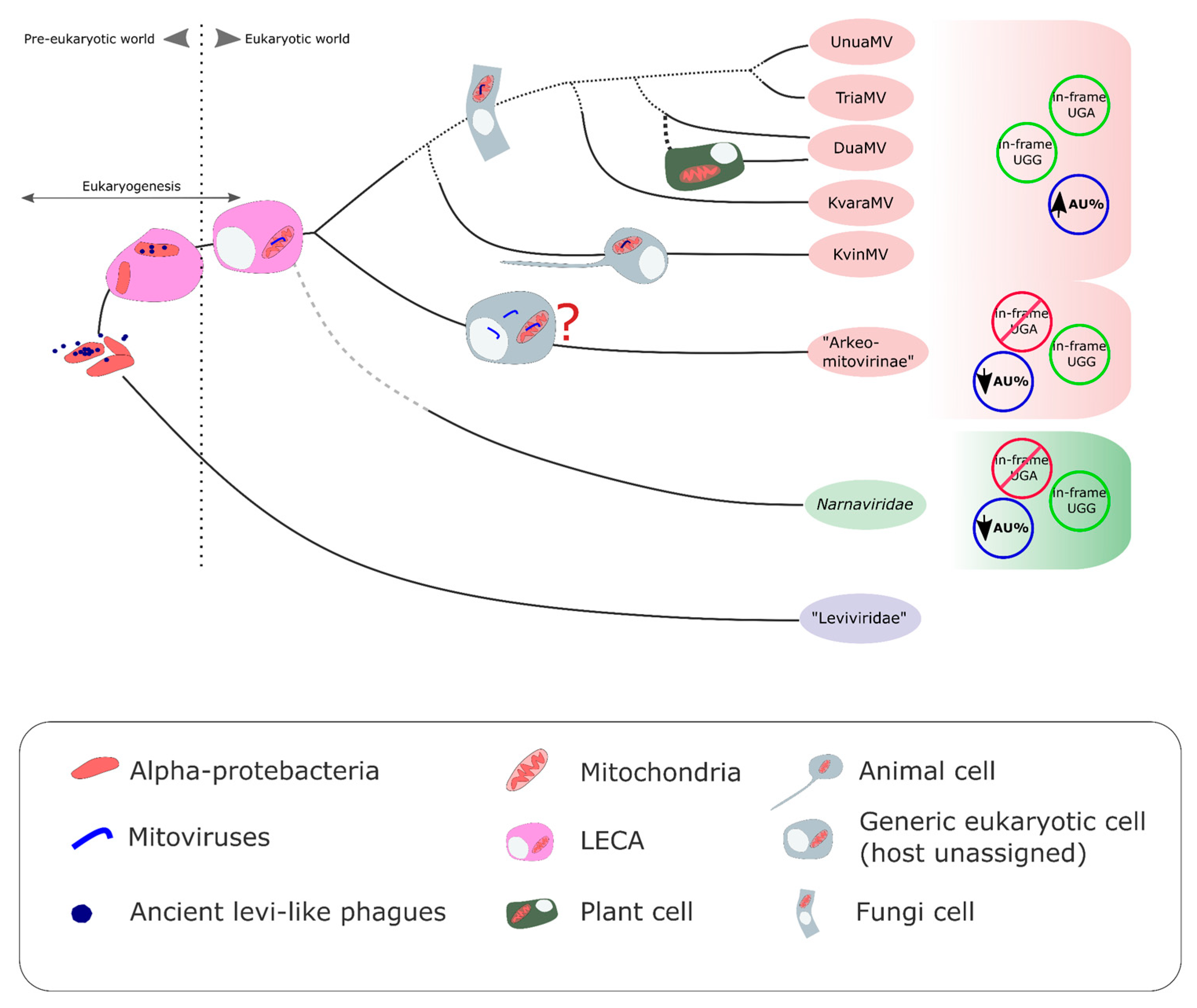
3.5. On the Origin of the New Putative Mitoviruses without In-Frame UGA(W) Codons
3.6. A Coherent Picture of the Evolution of Mitoviruses
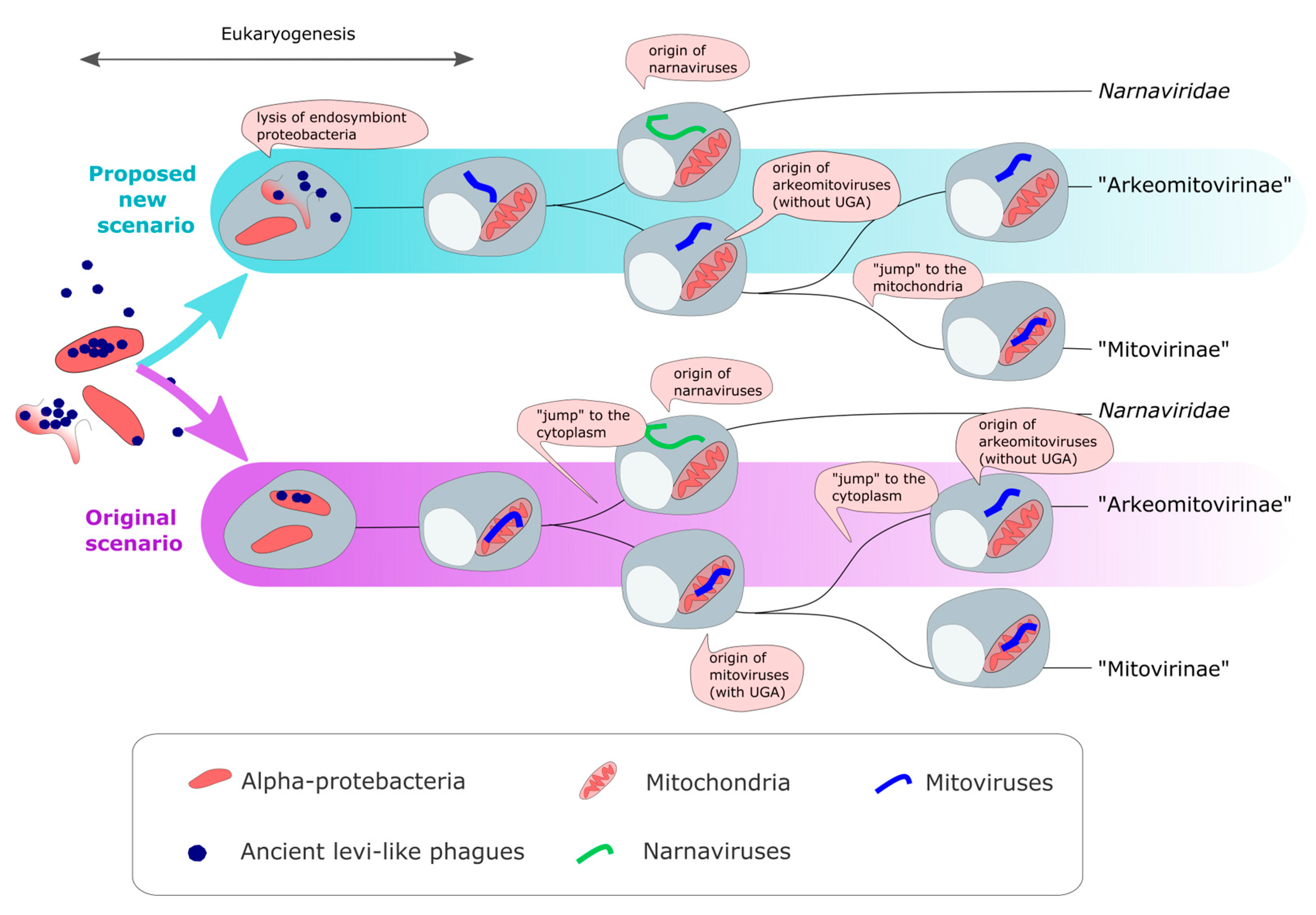
3.7. An Alternative Parsimonious Scenario for the Origins of Mitoviruses
4. Final Comments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hillman, B.I.; Cai, G. The Family Narnaviridae: Simplest of RNA viruses. Adv. Virus Res. 2013, 86, 149–176. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. Mbio 2018, 9, e02329-18. [Google Scholar] [CrossRef]
- Muñoz-Adalia, E.J.; Diez, J.J.; Fernández, M.M.; Hantula, J.; Vainio, E.J. Characterization of small RNAs originating from mitoviruses infecting the conifer pathogen Fusarium circinatum. Arch. Virol. 2018, 163, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Shahi, S.; Eusebio-Cope, A.; Kondo, H.; Hillman, B.I.; Suzuki, N. Investigation of Host Range of and Host Defense against a Mitochondrially Replicating Mitovirus. J. Virol. 2019, 93, e01503-18. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L. Mitovirus UGA(Trp) codon usage parallels that of host mitochondria. Virology 2017, 507, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef]
- Nerva, L.; Vigani, G.; Di Silvestre, D.; Ciuffo, M.; Forgia, M.; Chitarra, W.; Turina, M. Biological and Molecular Characterization of Chenopodium quinoa Mitovirus 1 Reveals a Distinct Small RNA Response Compared to Those of Cytoplasmic RNA Viruses. J. Virol. 2019, 93, e01998-18. [Google Scholar] [CrossRef]
- Fonseca, P.; Ferreira, F.; Da Silva, F.; Oliveira, L.S.; Marques, J.T.; Goes-Neto, A.; Aguiar, E.; Gruber, A. Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia longipalpis Using Genomic and Virus–Host Interaction Signatures. Viruses 2020, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A.; Warner, B.E.; Yerramsetty, P. Widespread mitovirus sequences in plant genomes. Peerj 2015, 3, e876. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, S.; Liu, L.; Cheng, J.; Fu, Y.; Jiang, D.; Xie, J. A mitovirus related to plant mitochondrial gene confers hypovirulence on the phytopathogenic fungus Sclerotinia sclerotiorum. Virus Res. 2015, 197, 127–136. [Google Scholar] [CrossRef]
- Katzourakis, A.; Gifford, R.J. Endogenous Viral Elements in Animal Genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Turina, M.; Ghignone, S.; Astolfi, N.; Silvestri, A.; Bonfante, P.; Lanfranco, L. The virome of the arbuscular mycorrhizal fungus Gigaspora margarita reveals the first report of DNA fragments corresponding to replicating non-retroviral RNA viruses in fungi. Environ. Microbiol. 2018, 20, 2012–2025. [Google Scholar] [CrossRef]
- Blair, C.D.; Olson, K.E.; Bonizzoni, M. The Widespread Occurrence and Potential Biological Roles of Endogenous Viral Elements in Insect Genomes. Curr. Issues Mol. Biol. 2020, 34, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Picarelli, M.A.S.C.; Forgia, M.; Rivas, E.B.; Nerva, L.; Chiapello, M.; Turina, M.; Colariccio, A. Extreme Diversity of Mycoviruses Present in Isolates of Rhizoctonia solani AG2-2 LP From Zoysia japonica From Brazil. Front. Cell. Infect. Microbiol. 2019, 9, 244. [Google Scholar] [CrossRef]
- Gilbert, C.; Belliardo, C. The diversity of endogenous viral elements in insects. Curr. Opin. Insect Sci. 2022, 49, 48–55. [Google Scholar] [CrossRef]
- Myers, J.M.; Bonds, A.E.; Clemons, R.A.; Thapa, N.A.; Simmons, D.R.; Carter-House, D.; Ortanez, J.; Liu, P.; Miralles-Durán, A.; Desirò, A.; et al. Survey of Early-Diverging Lineages of Fungi Reveals Abundant and Diverse Mycoviruses. Mbio 2020, 11, e02027-20. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, 1–21. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Release Executive Committee 53. Code: Proposal 2021.003F. July 2021. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 1 June 2022).
- Jacquat, A.G.; Ulla, S.B.; Debat, H.J.; Muñoz-Adalia, E.J.; Theumer, M.G.; Pedrajas, M.D.G.; Dambolena, J.S. An in silico analysis revealed a novel evolutionary lineage of putative mitoviruses. Environ. Microbiol. 2022, 24, 6463–6475. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, C.; Shi, M.; Buček, A.; Bourguignon, T.; Lo, N.; Holmes, E.C. Unmapped RNA Virus Diversity in Termites and Their Symbionts. Viruses 2020, 12, 1145. [Google Scholar] [CrossRef]
- Wille, M.; Harvey, E.; Shi, M.; Gonzalez-Acuña, D.; Holmes, E.C.; Hurt, A.C. Sustained RNA virome diversity in Antarctic penguins and their ticks. ISME J. 2020, 14, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J.; et al. Abundant and Diverse RNA Viruses in Insects Revealed by RNA-Seq Analysis: Ecological and Evolutionary Implications. Msystems 2020, 5, e00039-20. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Chen, Y.; Wei, X.; Cui, J. Viromes in marine ecosystems reveal remarkable invertebrate RNA virus diversity. Sci. China Life Sci. 2022, 65, 426–437. [Google Scholar] [CrossRef]
- Hirai, J.; Urayama, S.-I.; Takaki, Y.; Hirai, M.; Nagasaki, K.; Nunoura, T. RNA Virosphere in a Marine Zooplankton Community in the Subtropical Western North Pacific. Microbes Environ. 2022, 37, ME21066. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Grishin, N.V. PROMALS: Towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 2007, 23, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; Von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Hong, Y.; Dover, S.L.; Cole, T.E.; Brasier, C.M.; Buck, K.W. Multiple Mitochondrial Viruses in an Isolate of the Dutch Elm Disease FungusOphiostoma novo-ulmi. Virology 1999, 258, 118–127. [Google Scholar] [CrossRef]
- Sadiq, S.; Chen, Y.-M.; Zhang, Y.-Z.; Holmes, E.C. Resolving Deep Evolutionary Relationships within the RNA Virus Phylum Lenarviricota. Virus Evol. 2022, 8, veac055. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Sadiq, S.; Tian, J.-H.; Chen, X.; Lin, X.-D.; Shen, J.-J.; Chen, H.; Hao, Z.-Y.; Wille, M.; Zhou, Z.-C.; et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat. Microbiol. 2022, 7, 1312–1323. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Morris, T.J. Evolution and Taxonomy of Positive-Strand RNA Viruses: Implications of Comparative Analysis of Amino Acid Sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Garai, G. A review on multiple sequence alignment from the perspective of genetic algorithm. Genomics 2017, 109, 419–431. [Google Scholar] [CrossRef] [PubMed]
- DeRisi, J.L.; Huber, G.; Kistler, A.; Retallack, H.; Wilkinson, M.; Yllanes, D. An exploration of ambigrammatic sequences in narnaviruses. Sci. Rep. 2019, 9, 17982. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 2019, 17, 247–260. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Rodrigues, J.L.M.; Soudzilovskaia, N.A.; Barceló, M.; Olsson, P.A.; Song, C.; Tedersoo, L.; Yuan, F.; Yuan, F.; Lipson, D.A.; et al. Global biogeography of fungal and bacterial biomass carbon in topsoil. Soil Biol. Biochem. 2020, 151, 108024. [Google Scholar] [CrossRef]
- Yang, Y.; Lee, S.-H.; Jang, I.; Kang, H. Soil bacterial community structures across biomes in artificial ecosystems. Ecol. Eng. 2020, 158, 106067. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J. Gen. Virol. 2018, 99, 1331–1343. [Google Scholar] [CrossRef]
- Peay, K.; Kennedy, P.G.; Talbot, J.M. Dimensions of biodiversity in the Earth mycobiome. Nat. Rev. Genet. 2016, 14, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R.; Steidinger, B.S.; Bruns, T.D.; Peay, K.G. Competition–colonization tradeoffs structure fungal diversity. ISME J. 2018, 12, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements. Microbiol. Mol. Biol. Rev. 2014, 78, 278–303. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Albalat, R.; Cañestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef]
- Mills, D.R.; Peterson, R.L.; Spiegelman, S. An extracellular Darwinian experiment with a self-duplicating nucleic acid molecule. Proc. Natl. Acad. Sci. USA 1967, 58, 217–224. [Google Scholar] [CrossRef]
- Armougom, F.; Moretti, S.; Poirot, O.; Audic, S.; Dumas, P.; Schaeli, B.; Keduas, V.; Notredame, C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006, 34, W604–W608. [Google Scholar] [CrossRef]
- Holmes, E.C. What Does Virus Evolution Tell Us about Virus Origins? J. Virol. 2011, 85, 5247–5251. [Google Scholar] [CrossRef]
- Marz, M.; Beerenwinkel, N.; Drosten, C.; Fricke, M.; Frishman, D.; Hofacker, I.L.; Hoffmann, D.; Middendorf, M.; Rattei, T.; Stadler, P.F.; et al. Challenges in RNA virus bioinformatics. Bioinformatics 2014, 30, 1793–1799. [Google Scholar] [CrossRef]
- Neri, U.; Wolf, Y.I.; Roux, S.; Camargo, A.P.; Lee, B.; Kazlauskas, D.; Chen, I.M.; Ivanova, N.; Allen, L.Z.; Paez-Espino, D.; et al. Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell 2022, 185, 4023–4037. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquat, A.G.; Theumer, M.G.; Dambolena, J.S. Putative Mitoviruses without In-Frame UGA(W) Codons: Evolutionary Implications. Viruses 2023, 15, 340. https://doi.org/10.3390/v15020340
Jacquat AG, Theumer MG, Dambolena JS. Putative Mitoviruses without In-Frame UGA(W) Codons: Evolutionary Implications. Viruses. 2023; 15(2):340. https://doi.org/10.3390/v15020340
Chicago/Turabian StyleJacquat, Andrés Gustavo, Martín Gustavo Theumer, and José Sebastián Dambolena. 2023. "Putative Mitoviruses without In-Frame UGA(W) Codons: Evolutionary Implications" Viruses 15, no. 2: 340. https://doi.org/10.3390/v15020340
APA StyleJacquat, A. G., Theumer, M. G., & Dambolena, J. S. (2023). Putative Mitoviruses without In-Frame UGA(W) Codons: Evolutionary Implications. Viruses, 15(2), 340. https://doi.org/10.3390/v15020340