

Origin, Genetic Variation and Molecular Epidemiology of SARS-CoV-2 Strains Circulating in Sardinia (Italy) during the First and Second COVID-19 Epidemic Waves
 , ,
, ,  , , , , , , , , , , add
Show full author list
, , , , , , , , , , add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. Nucleic Acid Purification and Reverse Transcription Real-Time PCR
2.3. Whole-Genome Sequencing
2.3.1. Illumina Sequencing Method
2.3.2. Oxford Nanopore Technologies (ONT) Sequencing Method
2.4. Bioinformatic Analysis
2.5. Phylogenetic Analysis
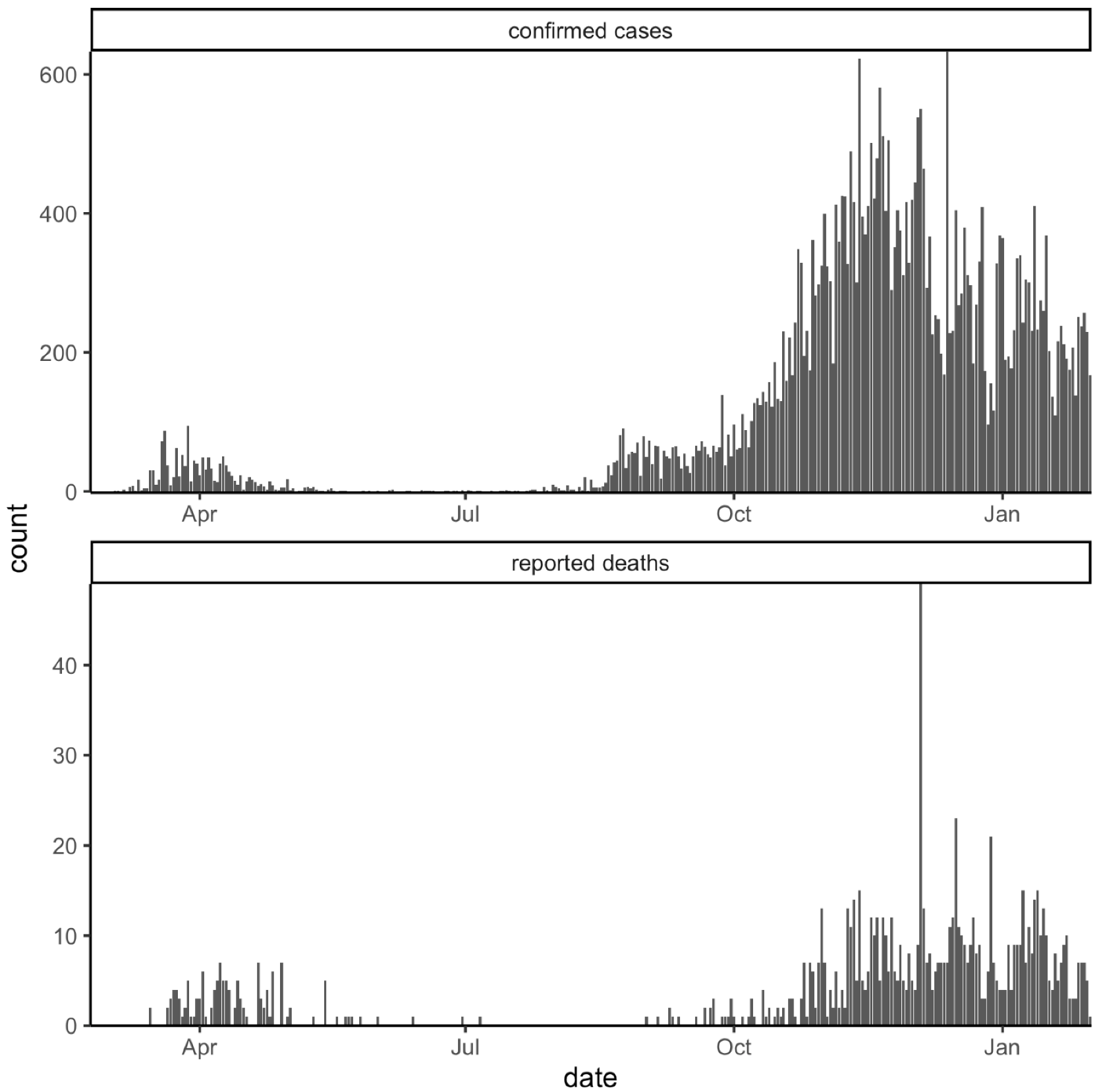
3. Results
3.1. Whole Genome Sequences Analysis
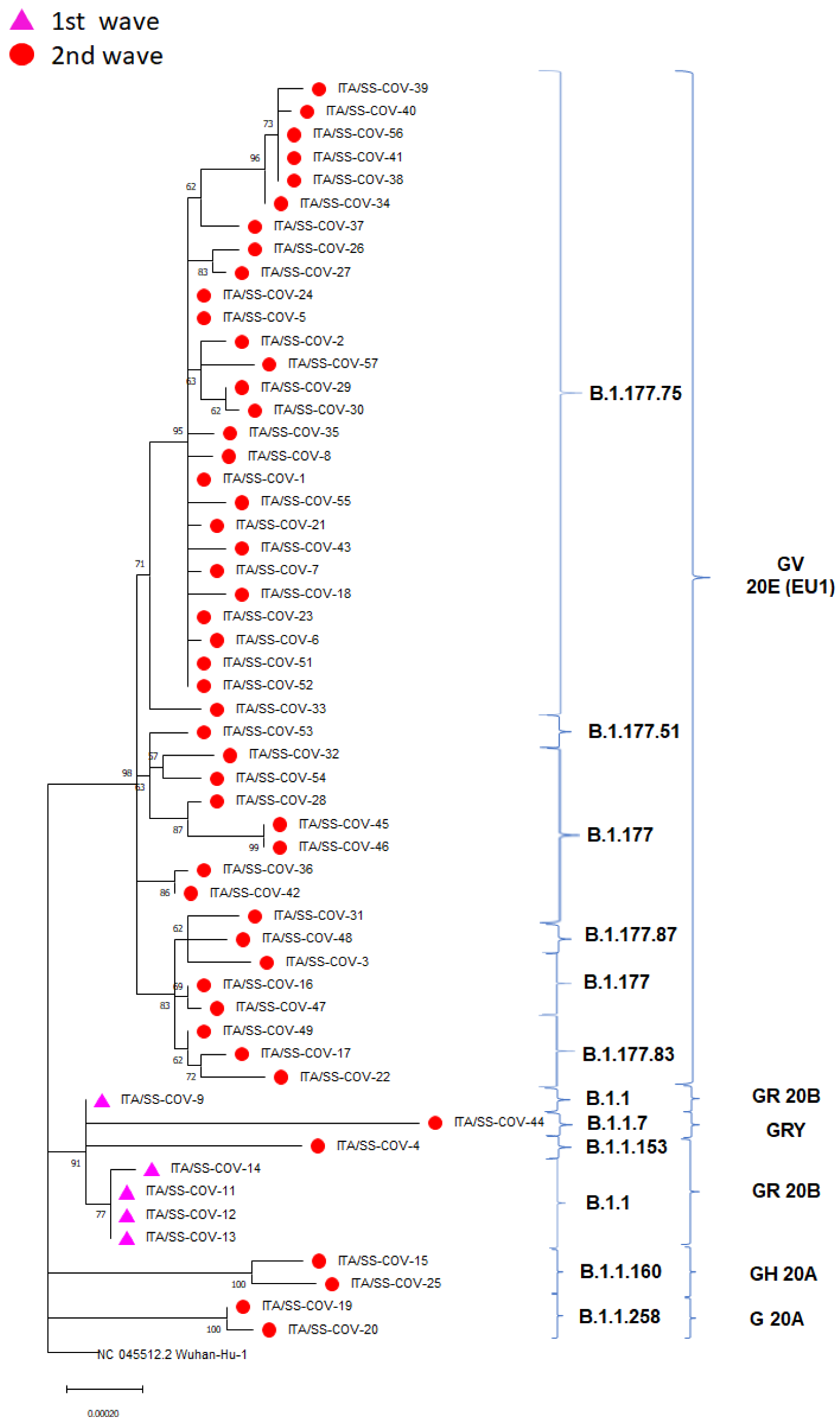
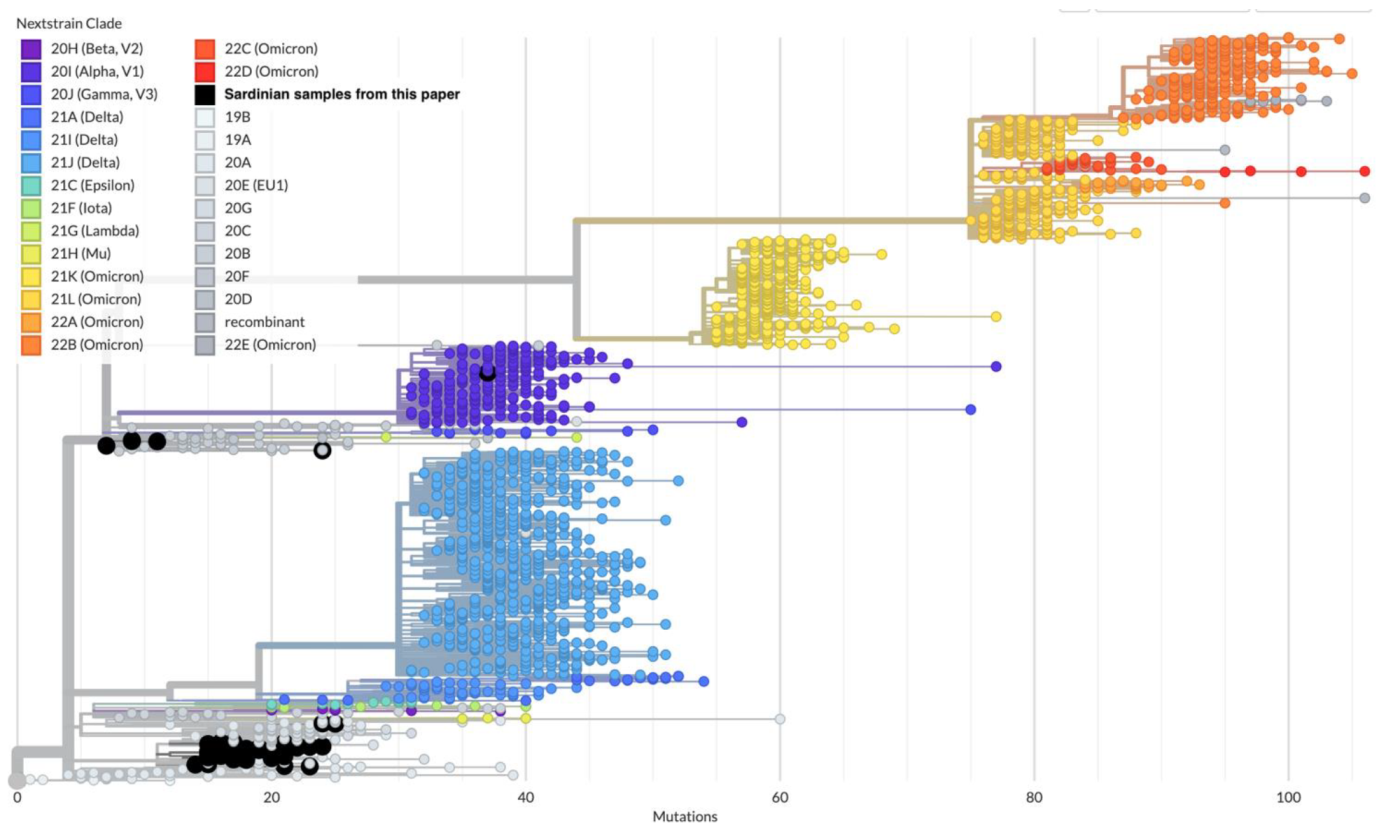
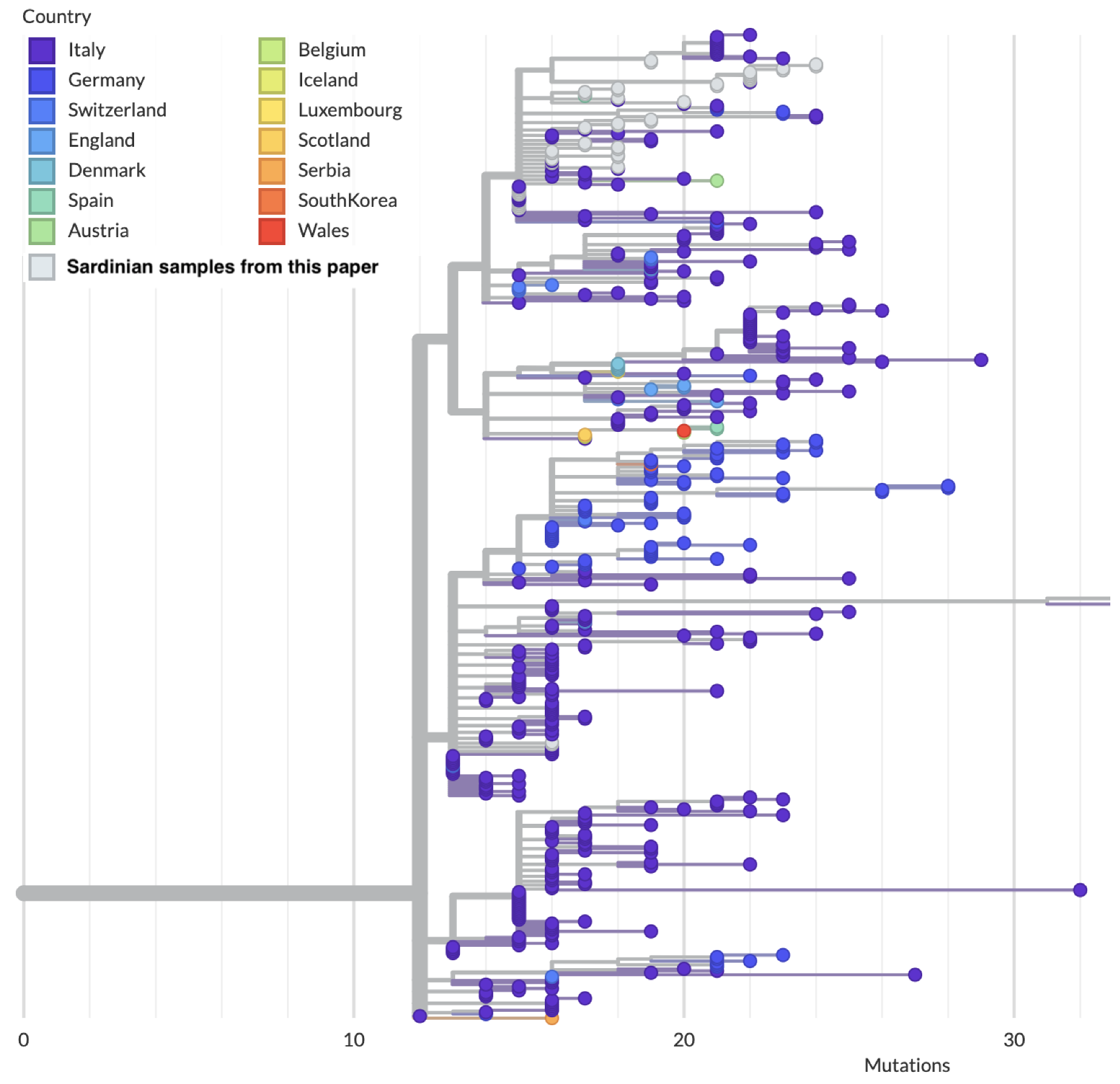
3.2. Phylogenetics and Molecular Epidemiology of the Outbreak
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Health Topics: Emerging Diseases; World Health Organization: Geneva, Switzerland; Available online: http://www.who.int/topics/emerging_diseases/en/ (accessed on 20 September 2022).
- Woolhouse, M.E.; Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 2005, 11, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Nii-Trebi, N.I. Emerging and Neglected Infectious Diseases: Insights, Advances, and Challenges. Biomed. Res. Int. 2017, 2017, 5245021. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Regional Office for South-East Asia, 2014. A Brief Guide to Emerging Infectious Diseases and Zoonoses. Available online: https://apps.who.int/iris/handle/10665/204722 (accessed on 20 September 2022).
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19-11-march-2020 (accessed on 23 March 2022).
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Zheng-Li, S. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 30 November 2021).
- Moubarak, M.; Kasozi, K.I.; Hetta, H.F.; Shaheen, H.M.; Rauf, A.; Al-Kuraishy, H.M.; Qusti, S.; Alshammari, E.M.; Ayikobua, E.T.; Ssempijja, F.; et al. The rise of SARS-CoV-2 variants and the role of convalescent plasma therapy for management of infections. Life 2021, 23, 734. [Google Scholar] [CrossRef]
- Hinch, R.; Panovska-Griffiths, J.; Probert, W.J.M.; Ferretti, L.; Wymant, C.; Di Lauro, F.; Baya, N.; Ghafari, M.; Abeler-Dörner, L.; COVID-19 Genomics UK (COG-UK) Consortium; et al. Estimating SARS-CoV-2 variant fitness and the impact of interventions in England using statistical and geo-spatial agent-based models. Philos. Trans. A Math. Phys. Eng. Sci. 2022, 380, 20210304. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Puci, M.V.; Loi, F.; Ferraro, O.E.; Cappai, S.; Rolesu, S.; Montomoli, C. COVID-19 Trend Estimation in the Elderly Italian Region of Sardinia. Front. Public Health 2020, 8, 153. [Google Scholar] [CrossRef]
- Puci, M.V.; Nosari, G.; Loi, F.; Puci, G.V.; Montomoli, C.; Ferraro, O.E. Risk Perception and Worries among Health Care Workers in the COVID-19 Pandemic: Findings from an Italian Survey. Healthcare 2020, 8, 535. [Google Scholar] [CrossRef]
- COVID-19 Data Repository by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University. Available online: https://github.com/CSSEGISandData/COVID-19 (accessed on 30 November 2021).
- InfluNET: Sistema di Sorveglianza Integrata dell’Influenza. Available online: https://w3.iss.it/site/rmi/influnet/pagine/stagioni.aspx (accessed on 10 January 2023).
- Ferraro, O.E.; Puci, M.V.; Montomoli, C.; Rolesu, S.; Cappai, S.; Loi, F. Official Data and Analytical Forecasts: Differences and Similarities Among Coronavirus Disease (COVID-19) Confirmed Cases and Deaths. Front. Med. 2020, 7, 239. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; Du Plessis, L.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.-Y.; et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 9263, 582–587. [Google Scholar] [CrossRef]
- Malune, P.; Piras, G.; Monne, M.; Fiamma, M.; Asproni, R.; Fancello, T.; Manai, A.; Carta, F.; Pira, G.; Fancello, P.; et al. Molecular Characterization of Severe Acute Respiratory Syndrome Coronavirus 2 Isolates from Central Inner Sardinia. Front. Microbiol. 2022, 12, 4335. [Google Scholar] [CrossRef]
- Gugliotta, C.; Gentili, D.; Marras, S.; Dettori, M.; Muglia, P.P.; Desole, M.G.; Acciaro, M.; Bellu, S.; Azara, A.; Castiglia, P. SARS-CoV-2 Epidemics in Retirement and Nursing Homes in Italy: A New Preparedness Assessment Model after the First Epidemic Wave. Int. J. Environ. Res. Public Health 2021, 18, 5712. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). Archive of Data for the Maps in Support of the Council Recommendation on a Coordinated Approach to Travel Measures in the EU. Available online: https://www.ecdc.europa.eu/en/publications-data/archive-data-maps-support-council-recommendation-coordinated-approach-travel (accessed on 19 September 2022).
- Decreto del Presidente del Consiglio dei Ministri, Dpcm del 3 Dicembre 2020. Available online: https://www.governo.it/sites/new.governo.it/files/dpcm_20201203.pdf (accessed on 10 January 2023).
- LoCost. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 12 December 2022).
- TrimGalore. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 10 March 2021).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef]
- Picard-Tools. Available online: https://github.com/broadinstitute/picard (accessed on 30 June 2021).
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing; Cornell University Press: Ithaca, NY, USA, 2012. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- GISAID. Available online: https://gisaid.org/database-features/covsurver-mutations-app/ (accessed on 10 May 2021).
- ARTICnetwork. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 30 May 2021).
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Sym. Ser. 1999, 41, 95–98. [Google Scholar]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 7, 1253–1256. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Phylogenetic Assignment Named Global Outbreak LINeages. Available online: https://github.com/hCoV-2019/pangolin (accessed on 30 May 2021).
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef]
- UCSC Genome Browser. Available online: https://genome.ucsc.edu/cgi-bin/hgPhyloPlace (accessed on 7 November 2022).
- AUSPICE. Available online: https://auspice.us (accessed on 7 November 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Ziebuhr, J. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 2005, 287, 57–94. [Google Scholar] [CrossRef]
- Chen, L.-D.; Zhang, Z.-Y.; Wei, X.-J.; Cai, Y.-Q.; Yao, W.-Z.; Wang, M.-H.; Huang, Q.-F.; Zhang, X.-B. Association between cytokine profiles and lung injury in COVID-19 pneumonia. Respir Res. 2020, 21, 201. [Google Scholar] [CrossRef]
- Hodcroft, H.B.; Zuber, M.; Nadeau, S.; Comas, I.; Candelas, F.G.; SeqCOVID-SPAIN Consortium; Stadler, T.; Neher, R.A. Spread of a SARS-CoV-2 variant through Europe in summer 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; Filipe, A.D.S.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171.e20–1187.e20. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome, structura, replication and pathogenesis. J. Med. Virol 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Weisblum, I.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Kemp, S.A.; Harvey, W.T.; Datir, R.P.; Collier, D.A.; Ferreira, I.A.; Carabelli, A.M.; Gupta, R.K.; Meng, B. Recurrent emergence and transmission of a SARS-CoV-2 spike deletion H69/V70. BioRxiv 2021. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Ruktanonchai, N.; Hong, S.L.; Colizza, V.; Poletto, C.; Broeck, F.V.D.; Gill, M.S.; Ji, X.; Levasseur, A.; Munnink, B.B.O.; et al. Untangling introductions and persistence in COVID-19 resurgence in Europe. Nature 2021, 595, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group; Abbott, S.; Kucharski, A.J.; Funk, S. Estimating the overdispersion in COVID-19 transmission using outbreak sizes outside China. Wellcome Open Res. 2020, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Wang, W.; Gao, L.; Wang, Y.; Luo, K.; Ren, L.; Zhan, Z.; Chen, X.; Zhao, S.; Huang, Y.; et al. Transmission heterogeneities, kinetics, and controllability of SARS-CoV-2. Science 2020, 371, 6526. [Google Scholar] [CrossRef]
- Pekar, J.; Worobey, M.; Moshiri, M.; Scheffler, K.; Wertheim, J.O. Timing the SARS-CoV-2 index case in Hubei province. Science 2021, 372, 412–417. [Google Scholar] [CrossRef]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- The Guardian: How Sardinia Went from Safe Haven to COVID-19 Hotspot. Available online: https://www.theguardian.com/world/2020/sep/06/how-sardinia-went-from-safe-haven-to-covid-19-hotspot (accessed on 14 December 2022).
- The New York Times: Available online: Summer Jet-Setters Turned Sardinia into a Virus Hot-Spot. Available online: https://www.nytimes.com/2020/09/09/world/europe/coronavirus-sardinia-italy.html (accessed on 14 December 2022).
- Forbes: 3000 Billionaire Clubbers Flee Quarantine on Italian Island. Available online: https://www.forbes.com/sites/oliverwilliams1/2020/08/28/3000-billionaire-clubbers-flee-quarantine-on-italian-island/ (accessed on 14 December 2022).
- The Guardian: Italy Contacting Visitors to Sardinian Nightclub after COVID-19 Cluster. Available online: https://www.theguardian.com/world/2020/aug/27/italy-traces-visitors-to-sardinian-nightclub-after-covid-19-cluster (accessed on 14 December 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Age (Years) | Sex | Residence | Collection Date | GISAID Accession Number | |
|---|---|---|---|---|---|---|
| ITA/SS-COV-21 | under 12 | <1 | M | Ozieri | 2 November 2020 | EPI_ISL_9145916 |
| ITA/SS-COV-03 | 10 | F | Sassari | 16 November 2020 | EPI_ISL_9145899 | |
| ITA/SS-COV-20 | 10 | F | Olbia | 23 October 2020 | EPI_ISL_9145915 | |
| ITA/SS-COV-24 | 12 | F | Sassari | 2 November 2020 | EPI_ISL_9145919 | |
| ITA/SS-COV-15 | 13–18 | 13 | F | Olbia | 2 September 2020 | EPI_ISL_9145910 |
| ITA/SS-COV-16 | 13 | M | Arzachena | 2 September 2020 | EPI_ISL_9145911 | |
| ITA/SS-COV-01 | 17 | F | Sassari | 20 October 2020 | EPI_ISL_9145897 | |
| ITA/SS-COV-19 | 17 | M | Olbia | 16 October 2020 | EPI_ISL_9145914 | |
| ITA/SS-COV-35 | 17 | M | Sassari | 30 November 2020 | EPI_ISL_9145930 | |
| ITA/SS-COV-51 | 18 | M | Sassari | 2 November 2020 | EPI_ISL_9145945 | |
| ITA/SS-COV-08 | 20–29 | 21 | F | Sassari | 8 October 2020 | EPI_ISL_9145904 |
| ITA/SS-COV-05 | 23 | F | Sassari | 16 October 2020 | EPI_ISL_9145901 | |
| ITA/SS-COV-23 | 24 | M | Sassari | 7 October 2020 | EPI_ISL_9145918 | |
| ITA/SS-COV-49 | 24 | F | Olbia | 2 September 2020 | EPI_ISL_9145944 | |
| ITA/SS-COV-29 | 25 | M | Sassari | 27 November 2020 | EPI_ISL_9145924 | |
| ITA/SS-COV-44 | 28 | F | Sassari | 28 January 2021 | EPI_ISL_9145939 | |
| ITA/SS-COV-30 | 29 | M | Sassari | 27 November 2020 | EPI_ISL_9145925 | |
| ITA/SS-COV-33 | 30–39 | 31 | M | Sassari | 28 November 2020 | EPI_ISL_9145928 |
| ITA/SS-COV-45 | 32 | F | Ozieri | 28 January 2021 | EPI_ISL_9145940 | |
| ITA/SS-COV-31 | 33 | F | Sassari | 27 November 2020 | EPI_ISL_9145926 | |
| ITA/SS-COV-25 | 34 | M | Sassari | 25 November 2020 | EPI_ISL_9145920 | |
| ITA/SS-COV-27 | 40–49 | 40 | M | Ozieri | 27 November 2020 | EPI_ISL_9145922 |
| ITA/SS-COV-42 | 43 | M | Sassari | 22 December 2020 | EPI_ISL_9145937 | |
| ITA/SS-COV-04 | 46 | M | Olbia | 2 November 2020 | EPI_ISL_9145900 | |
| ITA/SS-COV-06 | 46 | F | Sassari | 4 November 2020 | EPI_ISL_9145902 | |
| ITA/SS-COV-22 | 47 | F | Olbia | 17 November 2020 | EPI_ISL_9145917 | |
| ITA/SS-COV-47 | 47 | M | Sassari | 2 September 2020 | EPI_ISL_9145942 | |
| ITA/SS-COV-02 | 49 | F | Sassari | 2 November 2020 | EPI_ISL_9145898 | |
| ITA/SS-COV-13 | 49 | M | Sassari | 15 April 2020 | EPI_ISL_9145908 | |
| ITA/SS-COV-07 | 50–59 | 50 | M | Sassari | 12 October 2020 | EPI_ISL_9145903 |
| ITA/SS-COV-36 | 51 | F | Sassari | 1 December 2020 | EPI_ISL_9145931 | |
| ITA/SS-COV-52 | 52 | M | Sassari | 2 November 2020 | EPI_ISL_9145946 | |
| ITA/SS-COV-53 | 52 | M | Olbia | 25 November 2020 | EPI_ISL_9145947 | |
| ITA/SS-COV-54 | 52 | F | Sassari | 3 December 2020 | EPI_ISL_9145948 | |
| ITA/SS-COV-12 | 53 | F | Sassari | 15 April 2020 | EPI_ISL_9145907 | |
| ITA/SS-COV-57 | 55 | F | Sassari | 3 December 2020 | EPI_ISL_9145951 | |
| ITA/SS-COV-43 | 56 | M | Sassari | 25 November 2020 | EPI_ISL_9145938 | |
| ITA/SS-COV-46 | 57 | F | Ozieri | 5 January 2021 | EPI_ISL_9145941 | |
| ITA/SS-COV-48 | 58 | M | Olbia | 2 September 2020 | EPI_ISL_9145943 | |
| ITA/SS-COV-56 | 59 | F | Sassari | 27 November 2020 | EPI_ISL_9145950 | |
| ITA/SS-COV-17 | 60–69 | 63 | M | Olbia | 7 September 2020 | EPI_ISL_9145912 |
| ITA/SS-COV-37 | 65 | F | Sassari | 2 December 2020 | EPI_ISL_9145932 | |
| ITA/SS-COV-32 | 68 | M | Sassari | 27 November 2020 | EPI_ISL_9145927 | |
| ITA/SS-COV-14 | 70–79 | 72 | F | Torralba | 26 May 2020 | EPI_ISL_9145909 |
| ITA/SS-COV-55 | 73 | M | Ozieri | 30 December 2020 | EPI_ISL_9145949 | |
| ITA/SS-COV-28 | 75 | M | Olbia | 26 November 2020 | EPI_ISL_9145923 | |
| ITA/SS-COV-34 | over 80 | 83 | F | Sassari | 27 November 2020 | EPI_ISL_9145929 |
| ITA/SS-COV-09 | 84 | M | Sassari | 28 April 2020 | EPI_ISL_9145905 | |
| ITA/SS-COV-40 | 86 | F | Sassari | 4 December 2020 | EPI_ISL_9145935 | |
| ITA/SS-COV-39 | 89 | F | Sassari | 4 December 2020 | EPI_ISL_9145934 | |
| ITA/SS-COV-38 | 91 | F | Sassari | 4 December 2020 | EPI_ISL_9145933 | |
| ITA/SS-COV-41 | 93 | F | Sassari | 4 December 2020 | EPI_ISL_9145936 | |
| ITA/SS-COV-18 | 95 | M | Olbia | 16 October 2020 | EPI_ISL_9145913 | |
| ITA/SS-COV-11 | 99 | F | Torralba | 6 May 2020 | EPI_ISL_9145906 | |
| ITA/SS-COV-26 | 100 | M | Ozieri | 27 November 2020 | EPI_ISL_9145921 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocchigiani, A.M.; Ferretti, L.; Ledda, A.; Di Nardo, A.; Floris, M.; Bonelli, P.; Loi, F.; Idda, M.L.; Angioi, P.P.; Zinellu, S.; et al. Origin, Genetic Variation and Molecular Epidemiology of SARS-CoV-2 Strains Circulating in Sardinia (Italy) during the First and Second COVID-19 Epidemic Waves. Viruses 2023, 15, 277. https://doi.org/10.3390/v15020277
Rocchigiani AM, Ferretti L, Ledda A, Di Nardo A, Floris M, Bonelli P, Loi F, Idda ML, Angioi PP, Zinellu S, et al. Origin, Genetic Variation and Molecular Epidemiology of SARS-CoV-2 Strains Circulating in Sardinia (Italy) during the First and Second COVID-19 Epidemic Waves. Viruses. 2023; 15(2):277. https://doi.org/10.3390/v15020277
Chicago/Turabian StyleRocchigiani, Angela Maria, Luca Ferretti, Alice Ledda, Antonello Di Nardo, Matteo Floris, Piero Bonelli, Federica Loi, Maria Laura Idda, Pier Paolo Angioi, Susanna Zinellu, and et al. 2023. "Origin, Genetic Variation and Molecular Epidemiology of SARS-CoV-2 Strains Circulating in Sardinia (Italy) during the First and Second COVID-19 Epidemic Waves" Viruses 15, no. 2: 277. https://doi.org/10.3390/v15020277
APA StyleRocchigiani, A. M., Ferretti, L., Ledda, A., Di Nardo, A., Floris, M., Bonelli, P., Loi, F., Idda, M. L., Angioi, P. P., Zinellu, S., Fiori, M. S., Bechere, R., Capitta, P., Coccollone, A., Coradduzza, E., Dettori, M. A., Fattaccio, M. C., Gallisai, E., Maestrale, C., ... Dei Giudici, S. (2023). Origin, Genetic Variation and Molecular Epidemiology of SARS-CoV-2 Strains Circulating in Sardinia (Italy) during the First and Second COVID-19 Epidemic Waves. Viruses, 15(2), 277. https://doi.org/10.3390/v15020277