

Omicron-BA.1 Dispersion Rates in Mexico Varied According to the Regional Epidemic Patterns and the Diversity of Local Delta Subvariants
 , , , , , , , , , , ,
, , , , , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
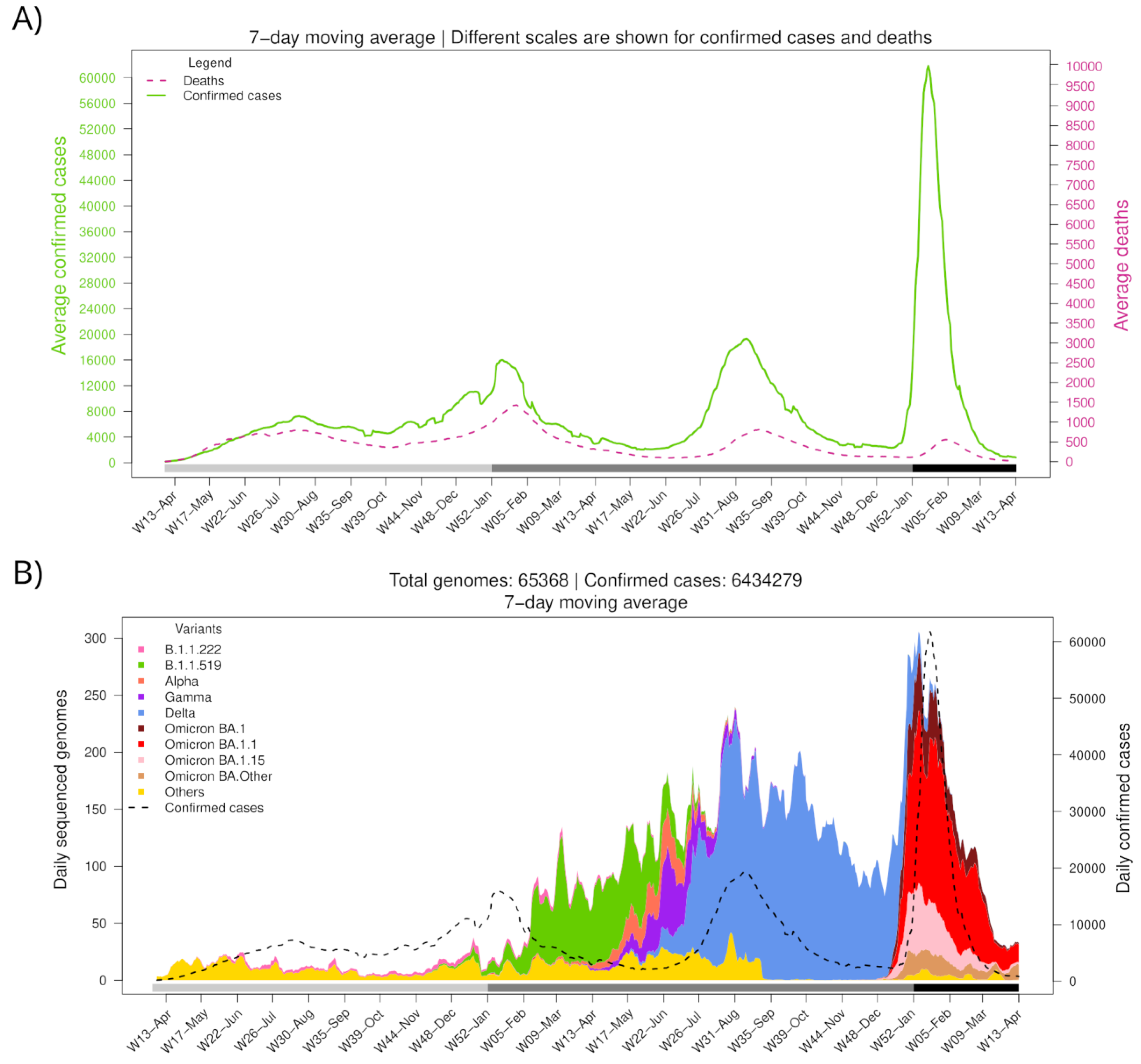
2.1. Epidemiological Information
2.2. Sequences Data Source
2.3. Lineage and Mutation Frequencies
2.4. Phylogeny and Haplotype Network
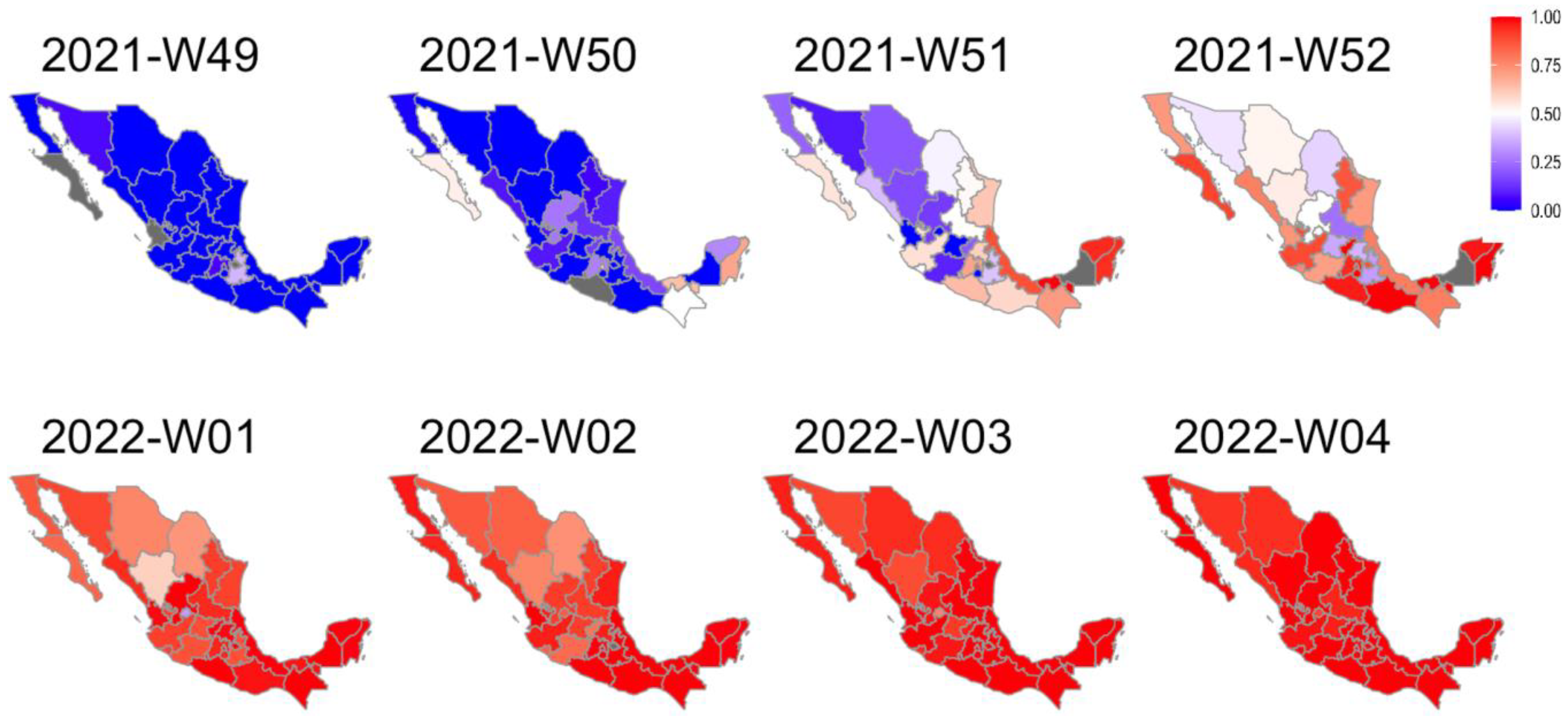
2.5. Analysis of the Regional Differences in the Introduction of BA.1.x and the Displacement of the Delta Variant
3. Results
3.1. BA.1.x Entry into Mexico
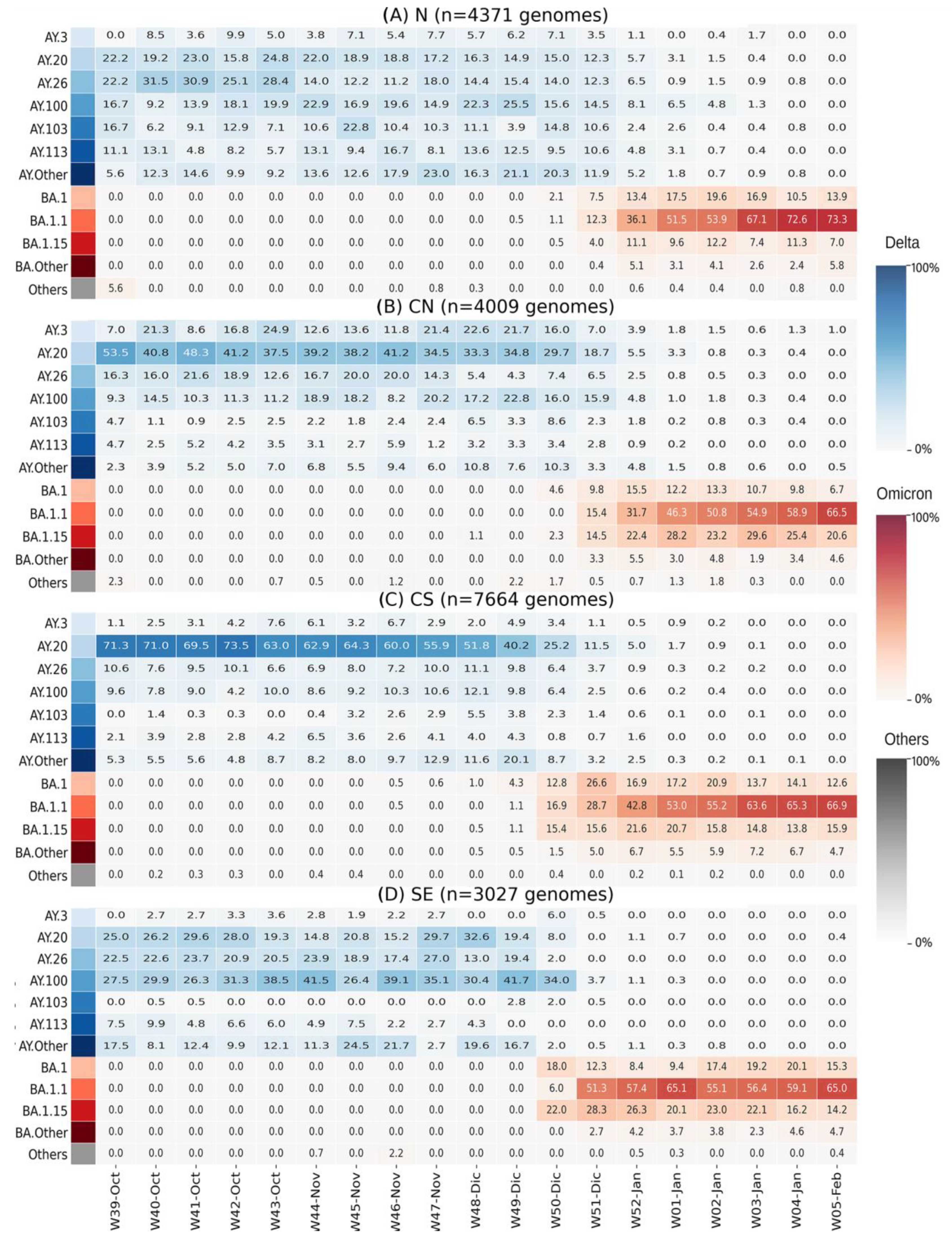
3.2. Diversity of BA.1.x in Mexico
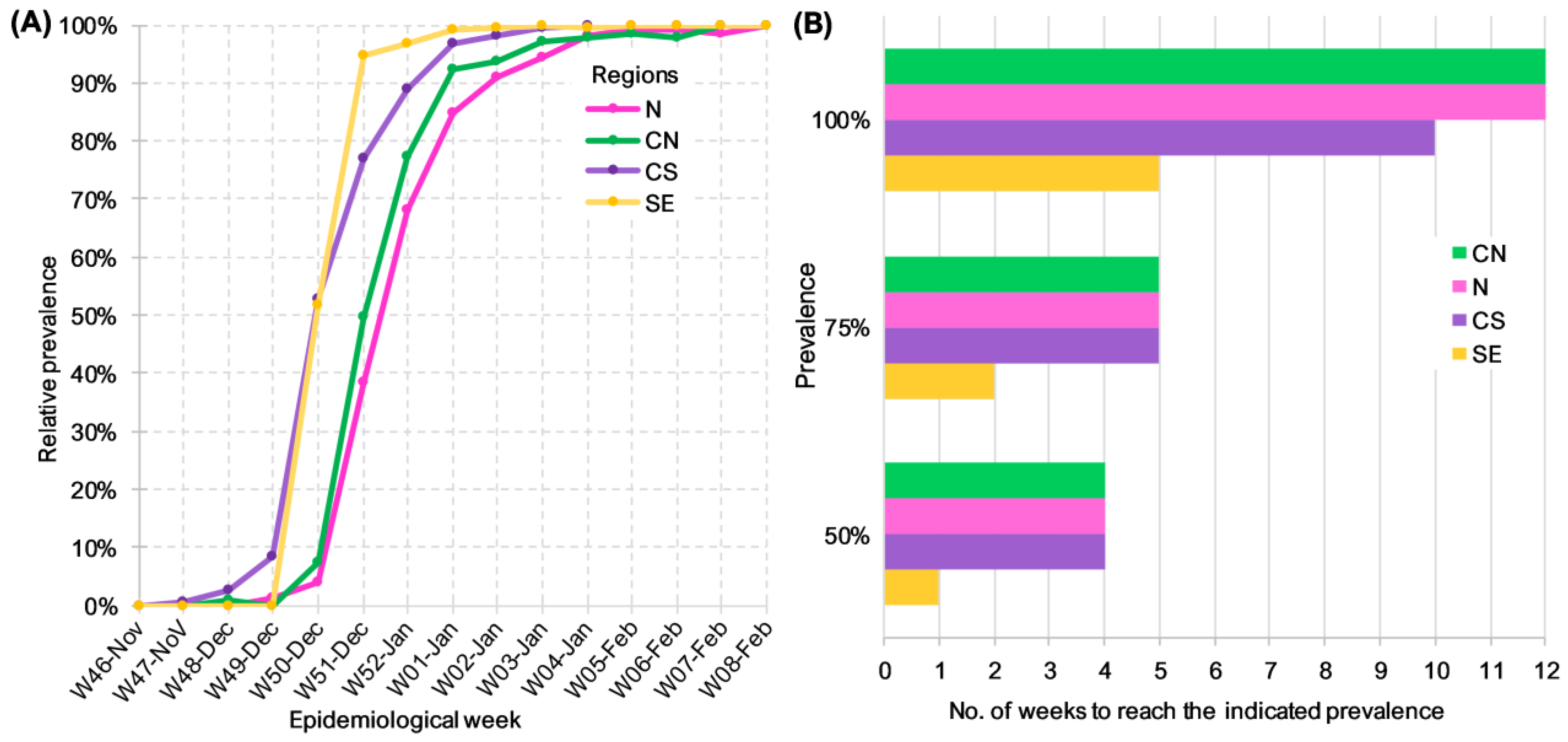
3.3. BA.1.x Entry in Mexico Was Asynchronous
3.4. Factors Affecting BA.1.x Dispersion Dynamics by Region
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salehi-Vaziri, M.; Fazlalipour, M.; Seyed Khorrami, S.M.; Azadmanesh, K.; Pouriayevali, M.H.; Jalali, T.; Shoja, Z.; Maleki, A. The Ins and Outs of SARS-CoV-2 Variants of Concern (VOCs). Arch. Virol. 2022, 167, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Tong, C.; Shi, W.; Zhang, A.; Shi, Z. Tracking and controlling the spatiotemporal spread of SARS-CoV-2 Omicron variant in South Africa. Travel Med. Infect. Dis. 2022, 46, 102252. [Google Scholar] [CrossRef]
- Grabowski, F.; Kochańczyk, M.; Lipniacki, T. The Spread of SARS-CoV-2 Variant Omicron with a Doubling Time of 2.0–3.3 Days Can Be Explained by Immune Evasion. Viruses 2022, 14, 294. [Google Scholar] [CrossRef]
- Noureddine, F.; Chakkour, M.; El Roz, A.; Reda, J.; Al Sahily, R.; Assi, A.; Joma, M.; Salami, H.; Hashem, S.; Harb, B.; et al. The Emergence of SARS-CoV-2 Variant(s) and Its Impact on the Prevalence of COVID-19 Cases in the Nabatieh Region, Lebanon. Med. Sci. 2021, 9, 40. [Google Scholar] [CrossRef]
- Taboada, B.; Zárate, S.; Iša, P.; Boukadida, C.; Vazquez-Perez, J.A.; Muñoz-Medina, J.E.; Ramírez-González, J.E.; Comas-García, A.; Grajales-Muñiz, C.; Rincón-Rubio, A.; et al. Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic. Viruses 2021, 13, 2161. [Google Scholar] [CrossRef]
- Bálint, G.; Vörös-Horváth, B.; Széchenyi, A. Omicron: Increased transmissibility and decreased pathogenicity. Signal Transduct. Target. Ther. 2022, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, P.; Trentini, F.; Petrone, D.; Mammone, A.; Ambrosio, L.; Manica, M.; Guzzetta, G.; d’Andrea, V.; Marziano, V.; Zardini, A.; et al. Tracking the Progressive Spread of the SARS-CoV-2 Omicron Variant in Italy, December 2021–January 2022. medRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Piantham, C.; Nishiura, H. Estimating relative generation times and reproduction numbers of Omicron BA.1 and BA.2 with respect to Delta variant in Denmark. Math. Biosci. Eng. 2022, 19, 9005–9017. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Takashita, E.; Kinoshita, N.; Yamayoshi, S.; Sakai-Tagawa, Y.; Fujisaki, S.; Ito, M.; Iwatsuki-Horimoto, K.; Chiba, S.; Halfmann, P.; Nagai, H.; et al. Efficacy of Antibodies and Antiviral Drugs against COVID-19 Omicron Variant. N. Engl. J. Med. 2022, 386, 995–998. [Google Scholar] [CrossRef]
- Brüssow, H. COVID-19: Omicron—The latest, the least virulent, but probably not the last variant of concern of SARS-CoV-2. Microb. Biotechnol. 2022, 15, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Ulloa, A.C.; Buchan, S.A.; Daneman, N.; Brown, K.A. Estimates of SARS-CoV-2 Omicron Variant Severity in Ontario, Canada. JAMA 2022, 327, 1286. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: A data linkage study. Lancet 2022, 399, 437–446. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.-W.; Hu, B.; Chai, Y.; Yuen, T.T.-T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.-C.; Liu, H.; et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Su, W.; Choy, K.T.; Gu, H.; Sia, S.F.; Cheng, K.M.; Nizami, S.I.N.; Krishnan, P.; Ng, Y.M.; Chang, L.D.J.; Liu, Y.; et al. Reduced Pathogenicity and Transmission Potential of Omicron BA.1 and BA.2 Sublineages Compared with the Early Severe Acute Respiratory Syndrome Coronavirus 2 D614G Variant in Syrian Hamsters. J. Infect. Dis. 2022, jiac276. [Google Scholar] [CrossRef]
- Du, X.; Tang, H.; Gao, L.; Wu, Z.; Meng, F.; Yan, R.; Qiao, S.; An, J.; Wang, C.; Qin, F.X.-F. Omicron adopts a different strategy from Delta and other variants to adapt to host. Signal Transduct. Target. Ther. 2022, 7, 45. [Google Scholar] [CrossRef]
- Yaxmehen Bello-Chavolla, O.; Eduardo Antonio-Villa, N.; Iván Valdés-Ferrer, S.; Fermín-Martínez, C.A.; Fernández-Chirino, L.; Ramírez-García, D.; Mancilla-Galindo, J.; Kammar-García, A.; Alberto Ávila-Funes, J.; Humberto Zúñiga-Gil, C.; et al. Effectiveness of a Nation-Wide COVID-19 Vaccination Program in Mexico. medRxiv 2022. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Taboada, B.; Zárate, S.; García-López, R.; Muñoz-Medina, J.E.; Sanchez-Flores, A.; Herrera-Estrella, A.; Boukadida, C.; Gómez-Gil, B.; Mojica, N.S.; Rosales-Rivera, M.; et al. Dominance of Three Sublineages of the SARS-CoV-2 Delta Variant in Mexico. Viruses 2022, 14, 1165. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Clement, M.J.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K.A. TCS: Estimating gene genealogies. In Proceedings of the 16th International Parallel and Distributed Processing Symposium, IPDPS 2002, Lauderdale, FL, USA, 15–19 April 2002; p. 184. [Google Scholar] [CrossRef]
- Chakkour, M.; Salami, A.; Olleik, D.; Kamal, I.; Noureddine, F.Y.; El Roz, A.; Ghssein, G. Risk Markers of COVID-19, a Study from South-Lebanon. Covid 2022, 2, 867–876. [Google Scholar] [CrossRef]
- Richter, J.; Koptides, D.; Tryfonos, C.; Alexandrou, D.; Christodoulou, C. Introduction, Spread and Impact of the SARS-CoV-2 Omicron Variants BA.1 and BA.2 in Cyprus. Microorganisms 2022, 10, 1688. [Google Scholar] [CrossRef]
- Lewnard, J.A.; Hong, V.X.; Patel, M.M.; Kahn, R.; Lipsitch, M.; Tartof, S.Y. Clinical outcomes associated with SARS-CoV-2 Omicron (B.1.1.529) variant and BA.1/BA.1.1 or BA.2 subvariant infection in Southern California. Nat. Med. 2022, 28, 1933–1943. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Y.; Zhao, Y.; He, D. Reduction in the infection fatality rate of Omicron variant compared with previous variants in South Africa. Int. J. Infect. Dis. 2022, 120, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.; Taboada, B.; Muñoz-Medina, J.E.; Iša, P.; Sanchez-Flores, A.; Boukadida, C.; Herrera-Estrella, A.; Mojica, N.S.; Rosales-Rivera, M.; Gómez-Gil, B.; et al. The Alpha Variant (B.1.1.7) of SARS-CoV-2 Failed to Become Dominant in Mexico. Microbiol. Spectr. 2022, 10, e0224021. [Google Scholar] [CrossRef] [PubMed]
- Cedro-Tanda, A.; Gómez-Romero, L.; de Anda-Jauregui, G.; Garnica-López, D.; Alfaro-Mora, Y.; Sánchez-Xochipa, S.; García-García, E.F.; Mendoza-Vargas, A.; Frías-Jiménez, E.J.; Moreno, B.; et al. Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City. Viruses 2022, 14, 545. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.F.; Cruz-Martins, N.; Luna, N.; Muñoz, M.; Ramírez, A.L.; Patiño, L.H.; Castañeda, S.A.; Ballesteros, N.; Ramírez, J.D. Genomic Diversity of SARS-CoV-2 Omicron Variant in South American Countries. Viruses 2022, 14, 1234. [Google Scholar] [CrossRef]
- Vauhkonen, H.; Nguyen, P.T.; Kant, R.; Plyusnin, I.; Erdin, M.; Kurkela, S.; Liimatainen, H.; Ikonen, N.; Blomqvist, S.; Liitsola, K.; et al. Introduction and Rapid Spread of SARS-CoV-2 Omicron Variant and Dynamics of BA.1 and BA.1.1 Sublineages, Finland, December 2021. Emerg. Infect. Dis. 2022, 28, 1229. [Google Scholar] [CrossRef]
- Linsenmeyer, K.; Gupta, K.; Madjarov, R.; Charness, M.E. Cryptic Transmission of the Delta Variant AY.3 Sublineage of SARS-CoV-2 among Fully Vaccinated Patients on an Inpatient Ward. medRxiv 2021. [Google Scholar] [CrossRef]
- Vest, A.; Houdeshell, H.; Clark, C.; Ghorpade, V.; Case, R.; Schrecker, J.; Hardison, M. eP333: Tracking the emergence of SARS-CoV-2 variants of concern in vaccinated and unvaccinated patients. Genet. Med. 2022, 24, S208–S209. [Google Scholar] [CrossRef]
- Ranjan, P.; Neha; Devi, C.; Devar, K.A.; Das, P. The influence of new SARS-CoV-2 variant Omicron (B.1.1.529) on vaccine efficacy, its correlation to Delta variants: A computational approach. Microb. Pathog. 2022, 169, 105619. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Mexico | USA | Canada | Brazil | South America | Rest of the Word |
|---|---|---|---|---|---|---|
| BA.1 | 16.8% | 12.1% | 38.3% | 37.5% | 24.9% | 16.2% |
| BA.1.1 | 57.6% | 52.1% | 13.4% | 27.4% | 57.0% | 19.0% |
| BA.1.1.18 | 1.2% | 7.4% | 0.7% | 0.1% | 0.3% | 0.1% |
| BA.1.15 | 17.1% | 14.0% | 7.3% | 6.2% | 8.2% | 2.0% |
| BA.1.17 | 1.2% | 0.6% | 1.3% | 0.9% | 1.4% | 2.6% |
| BA.1.20 | 1.4% | 3.5% | 2.4% | 0.1% | 0.5% | 0.2% |
| Others | 4.7% | 10.3% | 36.8% | 27.7% | 7.7% | 60.0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zárate, S.; Taboada, B.; Rosales-Rivera, M.; García-López, R.; Muñoz-Medina, J.E.; Sanchez-Flores, A.; Herrera-Estrella, A.; Gómez-Gil, B.; Selem Mojica, N.; Salas-Lais, A.G.; et al. Omicron-BA.1 Dispersion Rates in Mexico Varied According to the Regional Epidemic Patterns and the Diversity of Local Delta Subvariants. Viruses 2023, 15, 243. https://doi.org/10.3390/v15010243
Zárate S, Taboada B, Rosales-Rivera M, García-López R, Muñoz-Medina JE, Sanchez-Flores A, Herrera-Estrella A, Gómez-Gil B, Selem Mojica N, Salas-Lais AG, et al. Omicron-BA.1 Dispersion Rates in Mexico Varied According to the Regional Epidemic Patterns and the Diversity of Local Delta Subvariants. Viruses. 2023; 15(1):243. https://doi.org/10.3390/v15010243
Chicago/Turabian StyleZárate, Selene, Blanca Taboada, Mauricio Rosales-Rivera, Rodrigo García-López, José Esteban Muñoz-Medina, Alejandro Sanchez-Flores, Alfredo Herrera-Estrella, Bruno Gómez-Gil, Nelly Selem Mojica, Angel Gustavo Salas-Lais, and et al. 2023. "Omicron-BA.1 Dispersion Rates in Mexico Varied According to the Regional Epidemic Patterns and the Diversity of Local Delta Subvariants" Viruses 15, no. 1: 243. https://doi.org/10.3390/v15010243
APA StyleZárate, S., Taboada, B., Rosales-Rivera, M., García-López, R., Muñoz-Medina, J. E., Sanchez-Flores, A., Herrera-Estrella, A., Gómez-Gil, B., Selem Mojica, N., Salas-Lais, A. G., Vazquez-Perez, J. A., Cabrera-Gaytán, D. A., Fernandes-Matano, L., Uribe-Noguez, L. A., Chale-Dzul, J. B., Maldonado Meza, B. I., Mejía-Nepomuceno, F., Pérez-Padilla, R., Gutiérrez-Ríos, R. M., ... Arias, C. F. (2023). Omicron-BA.1 Dispersion Rates in Mexico Varied According to the Regional Epidemic Patterns and the Diversity of Local Delta Subvariants. Viruses, 15(1), 243. https://doi.org/10.3390/v15010243