

Comparative Analysis of Conformational Dynamics and Systematic Characterization of Cryptic Pockets in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB.1 Spike Complexes with the ACE2 Host Receptor: Confluence of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites



Abstract
:1. Introduction
2. Materials and Methods
2.1. All-Atom Molecular Dynamics Simulations
2.2. Markov State Model Analysis of the Omicron RBD-ACE2 Complexes
2.3. Coarse-Grained Dynamics Simulations of the Omicron S Trimer Complexes with ACE2
2.4. Mutational Scanning Analysis of the RBD-ACE2 Binding Interactions and Inter-Protomer Trimer Interactions
2.5. Machine Learning Detection of Cryptic Pockets
3. Results
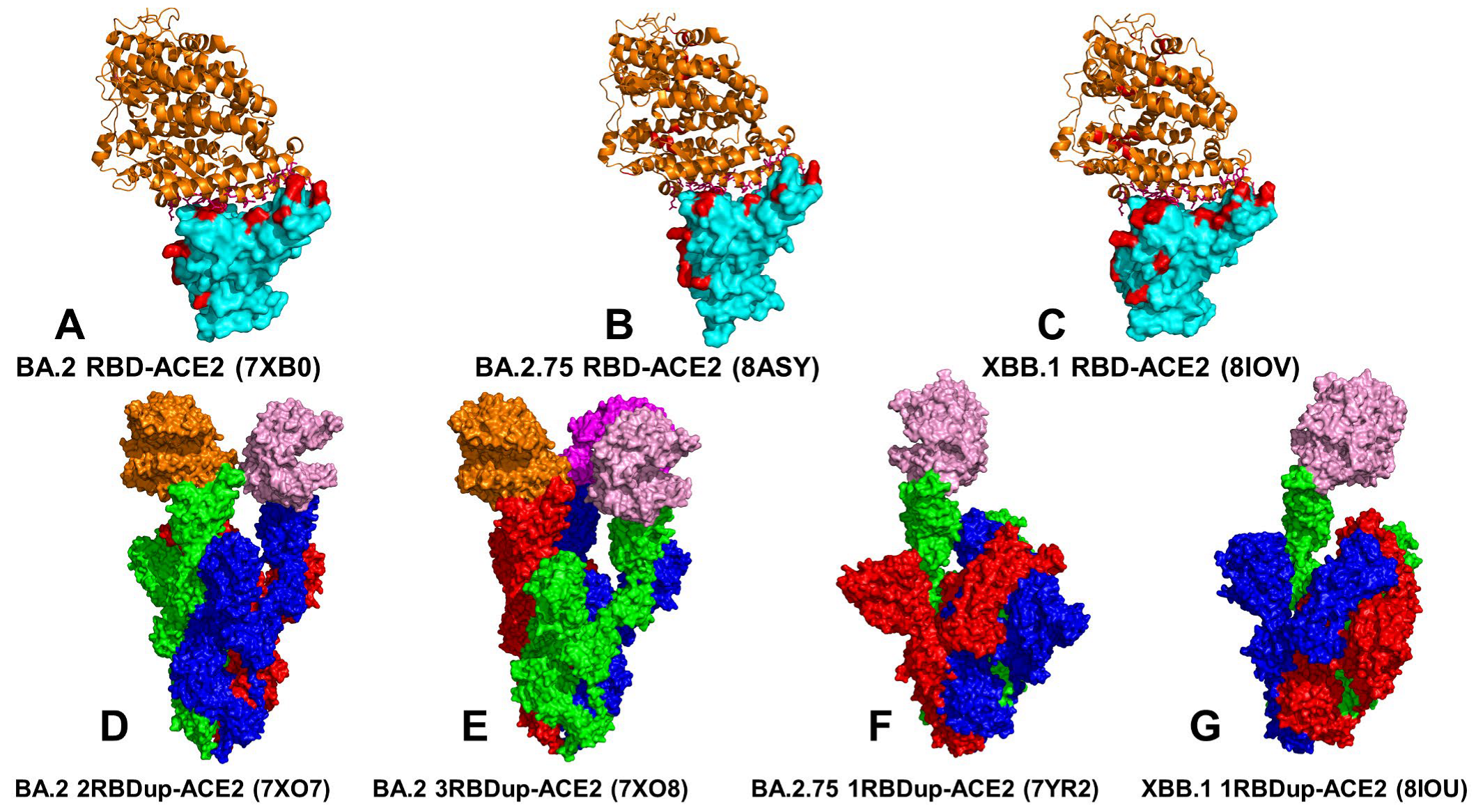
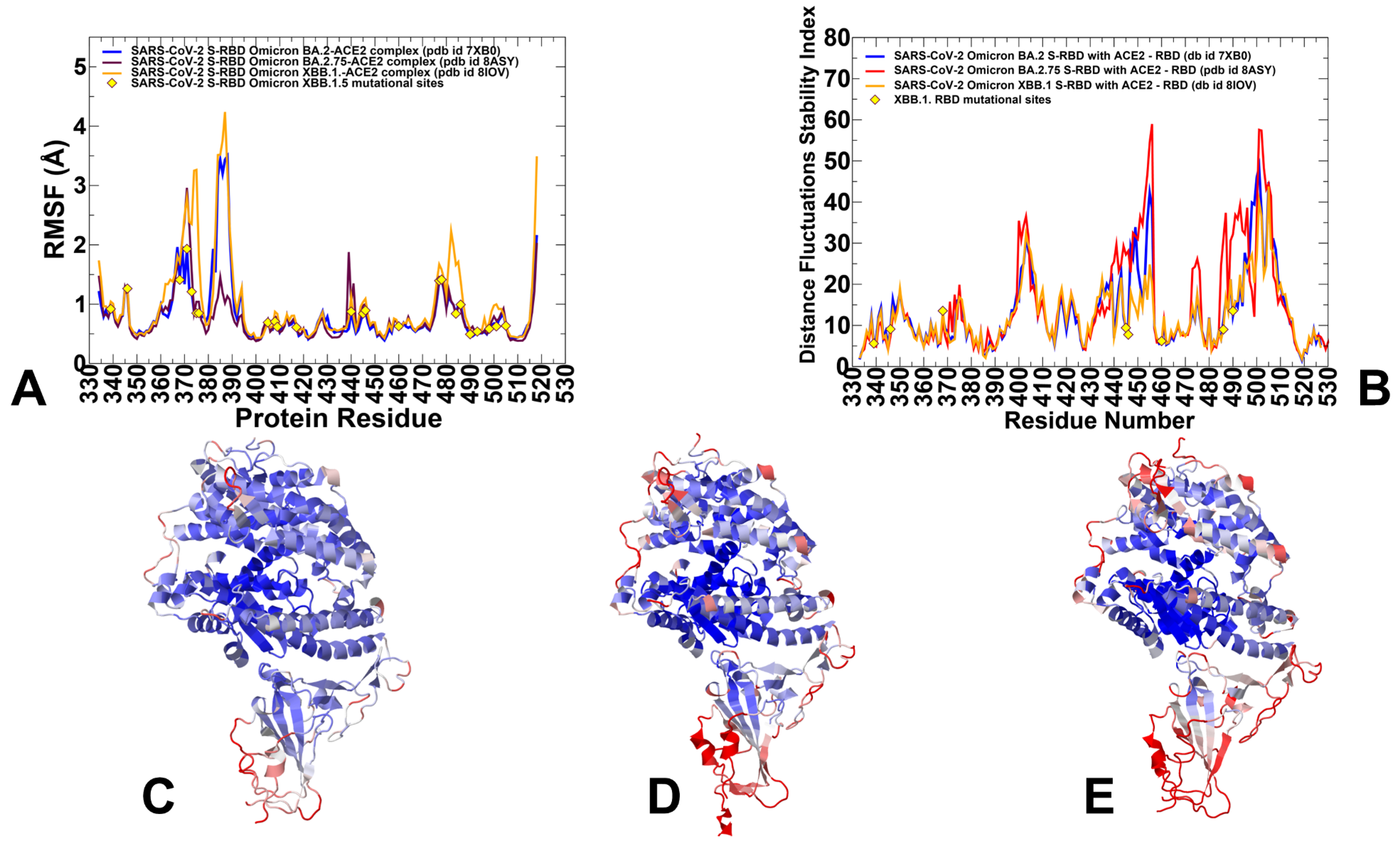
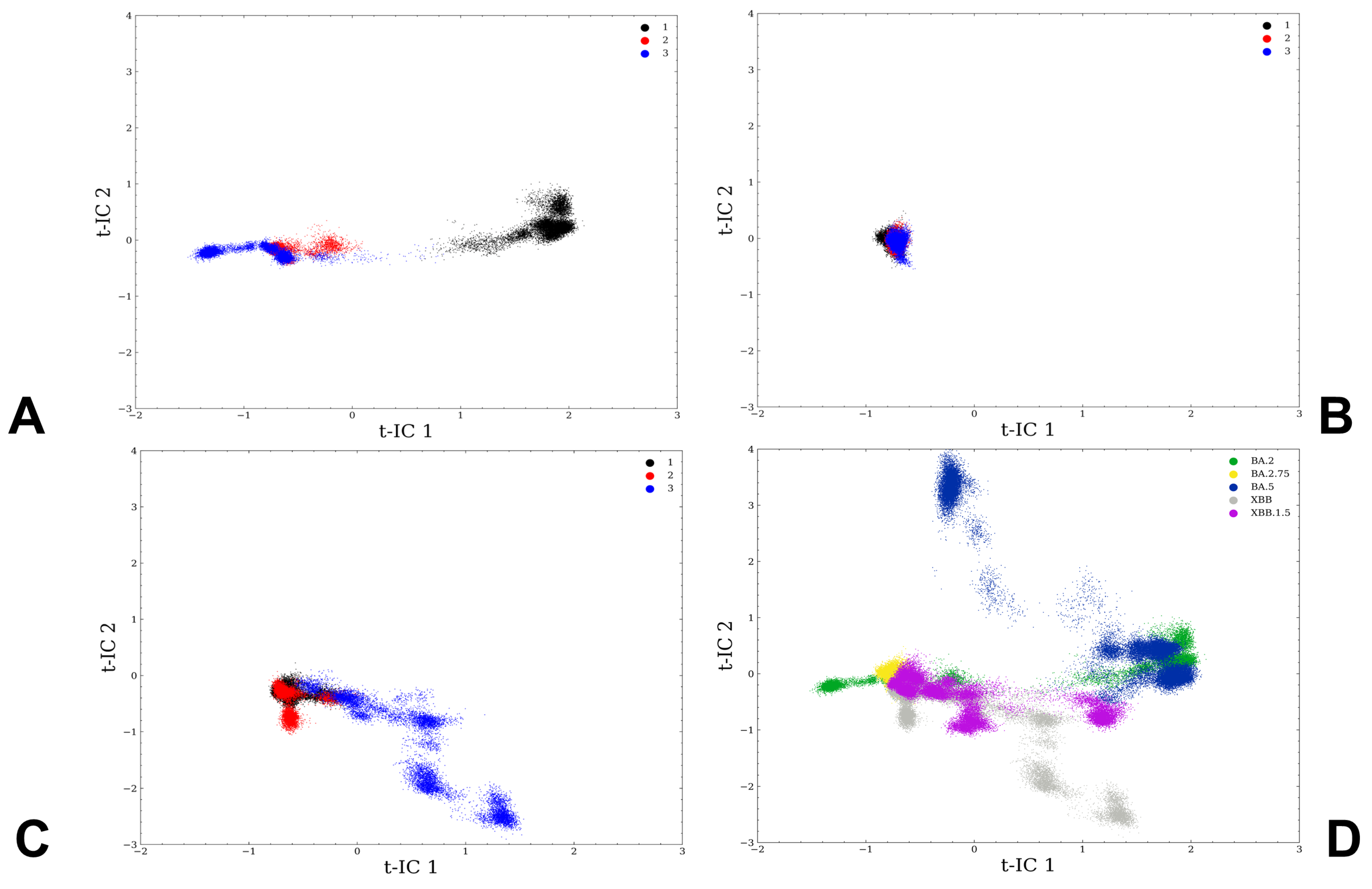
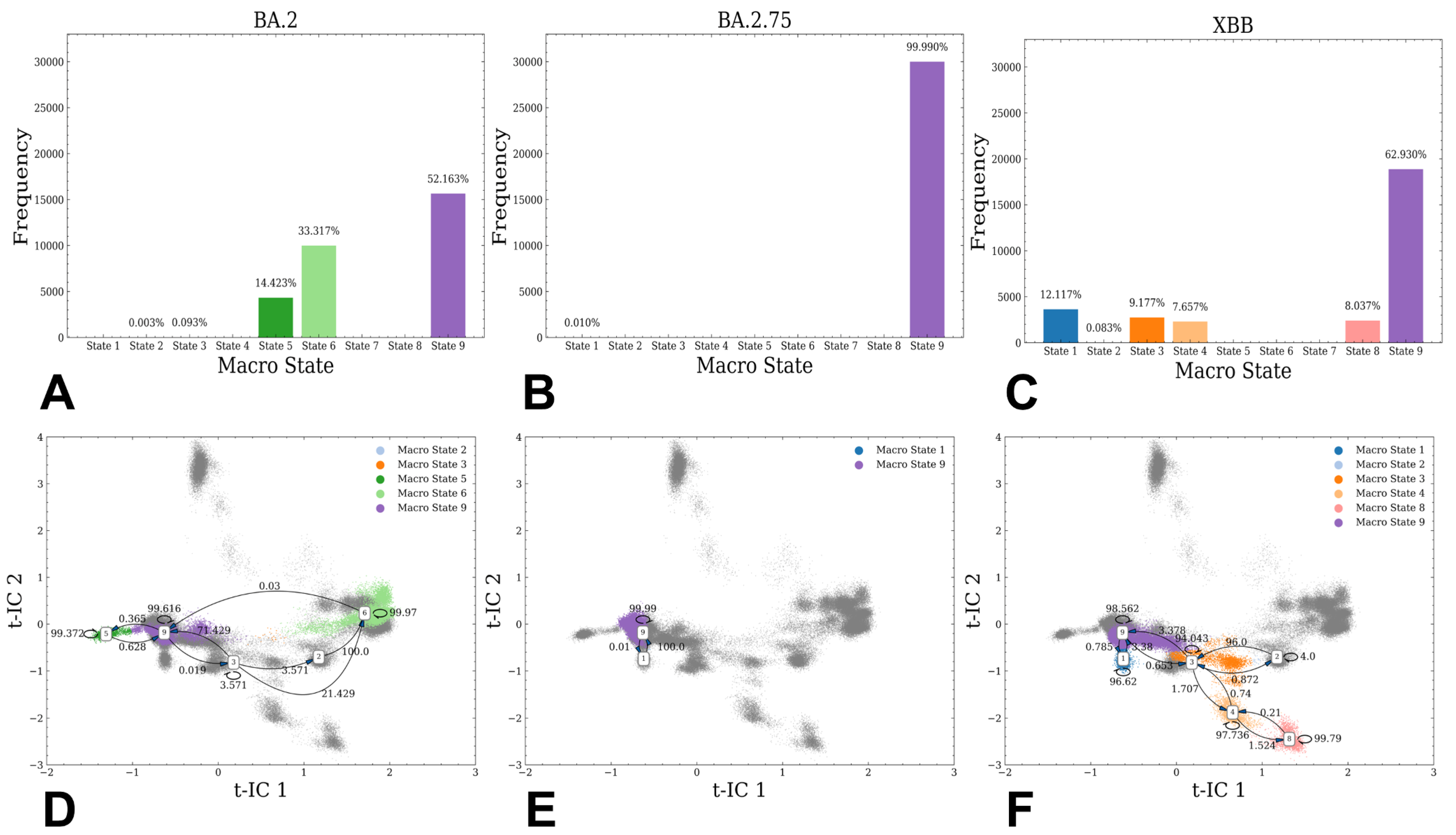
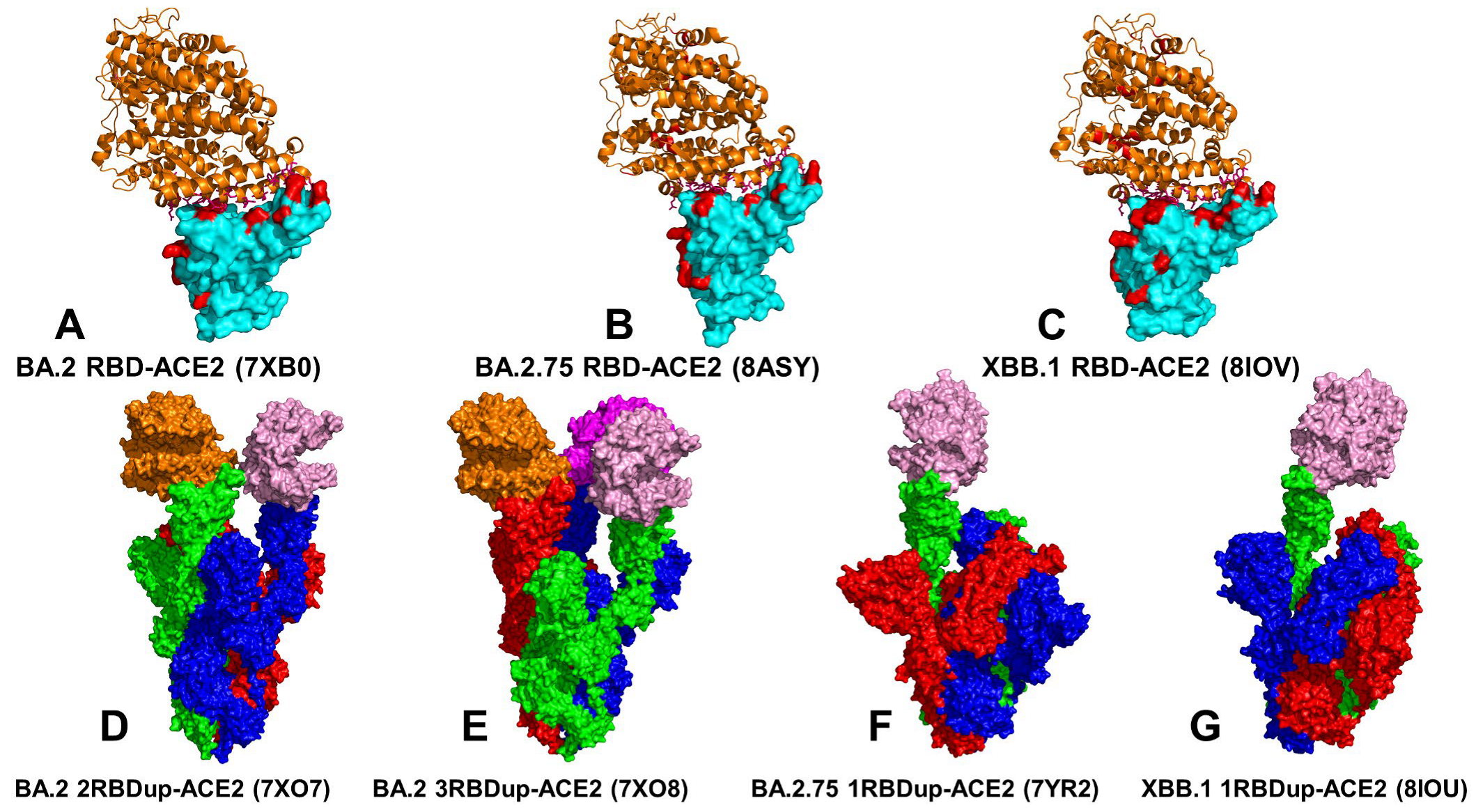
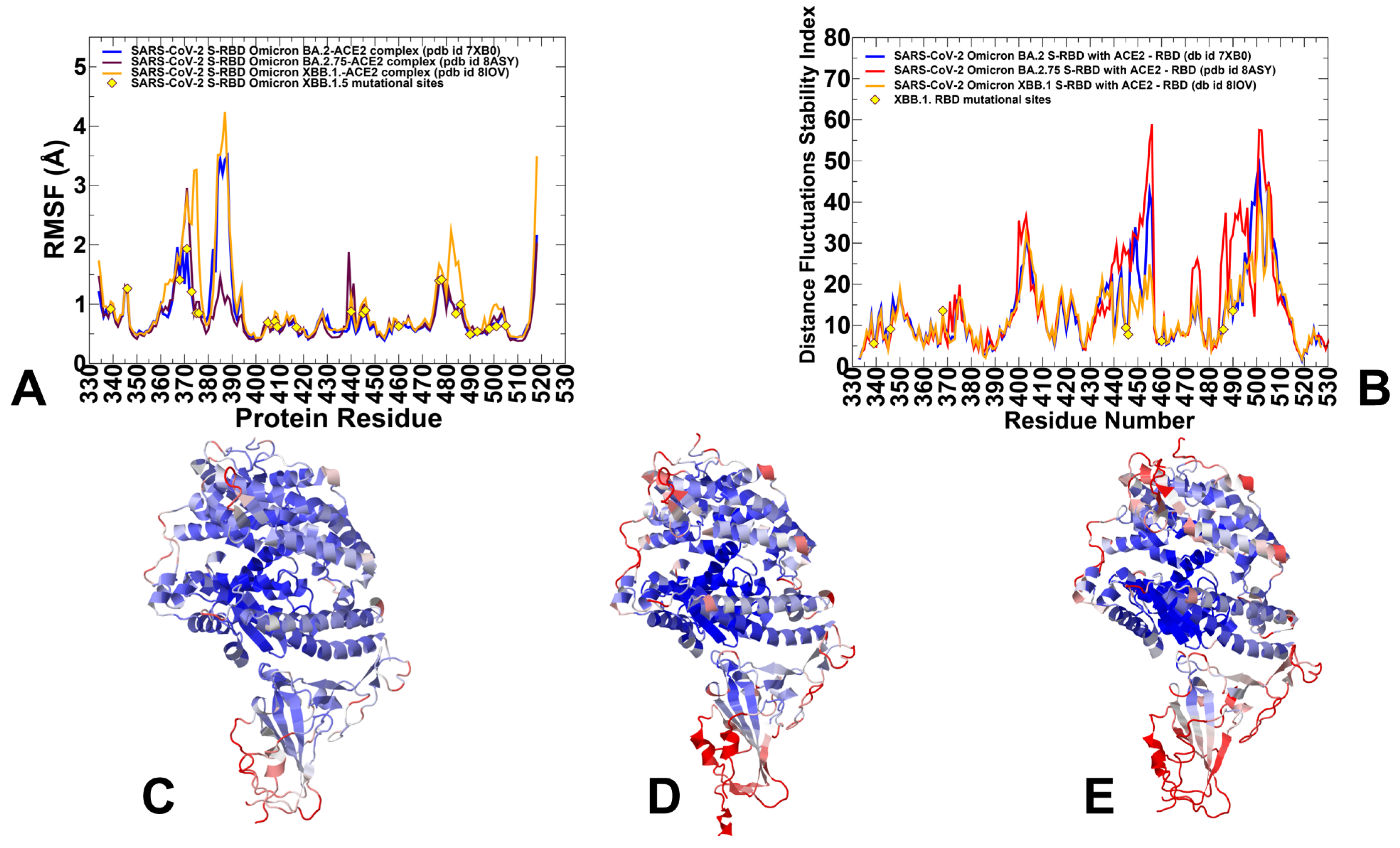
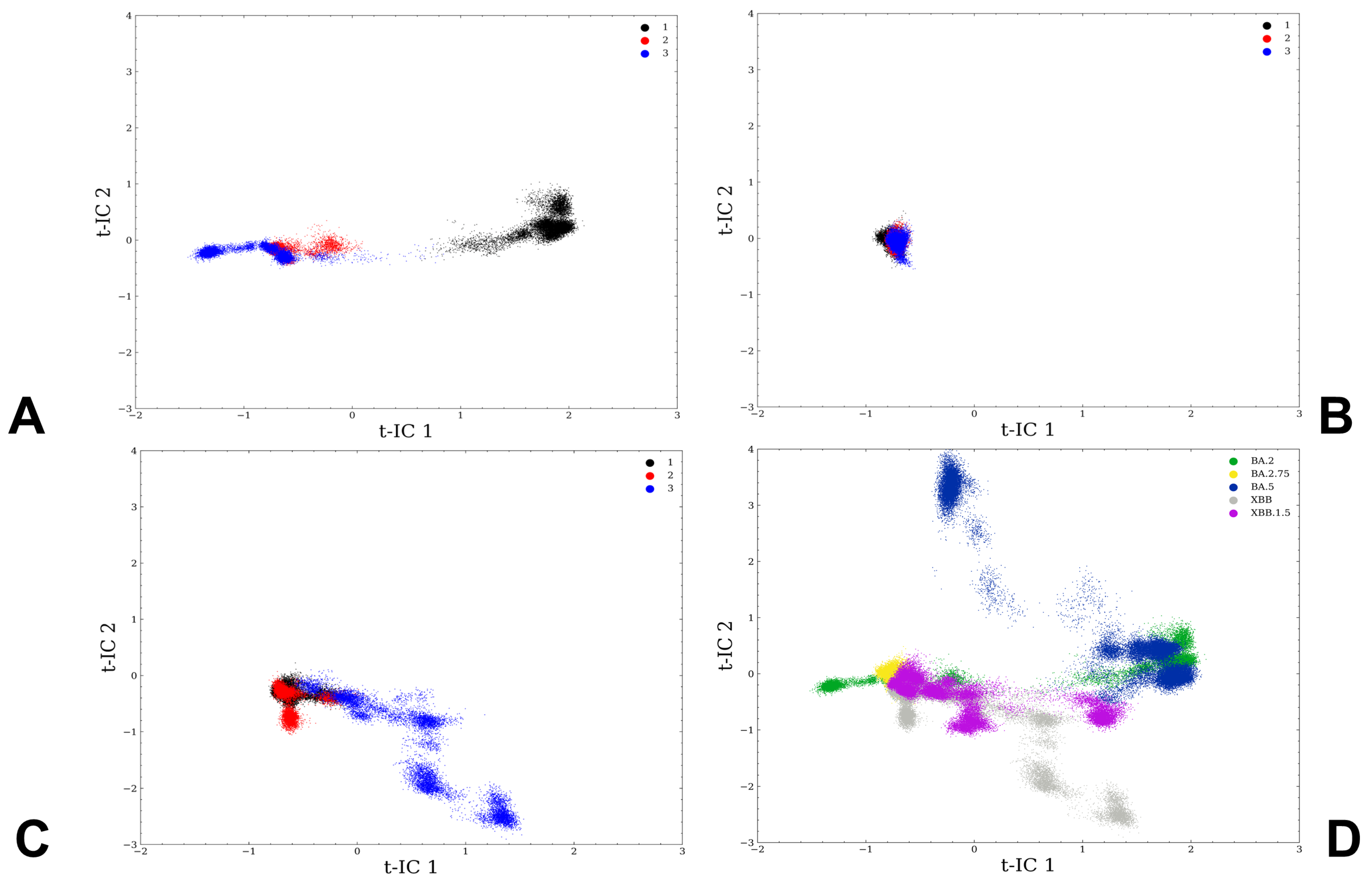
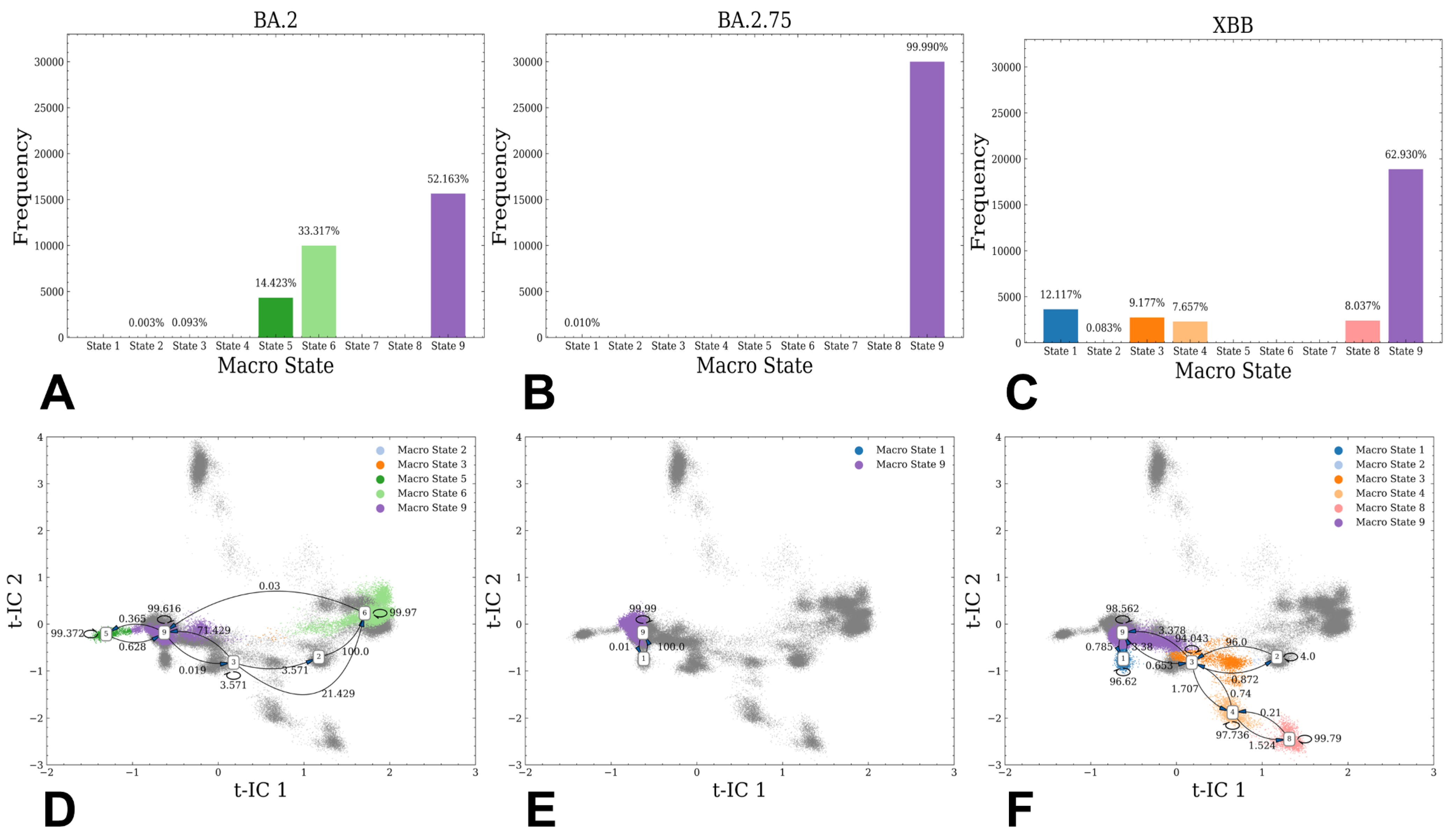
3.1. Atomistic MD Simulations and Markov State Model Analysis of the Omicron BA.2, BA.2.75 and XBB.1 RBD-ACE2 Complexes
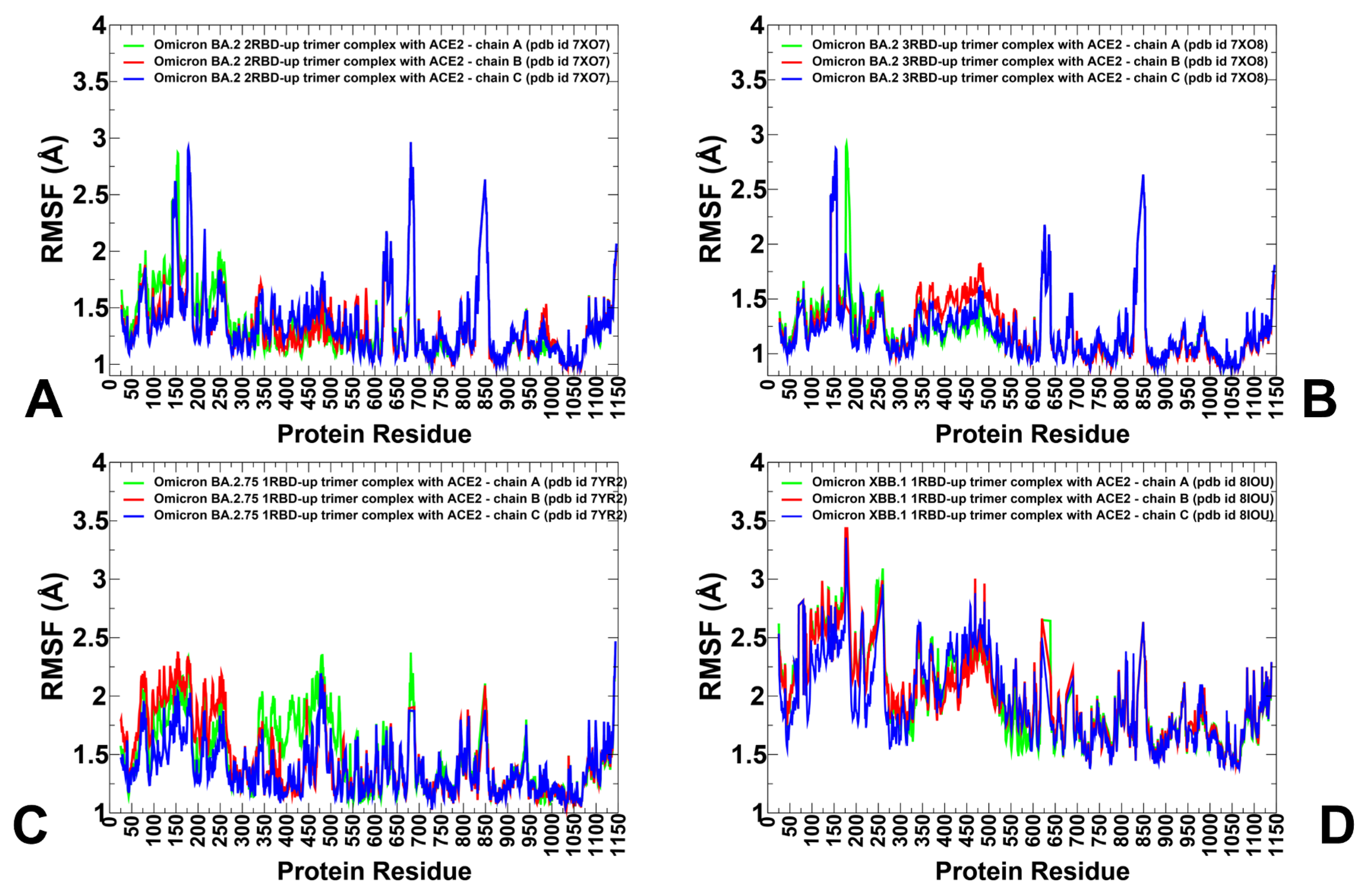
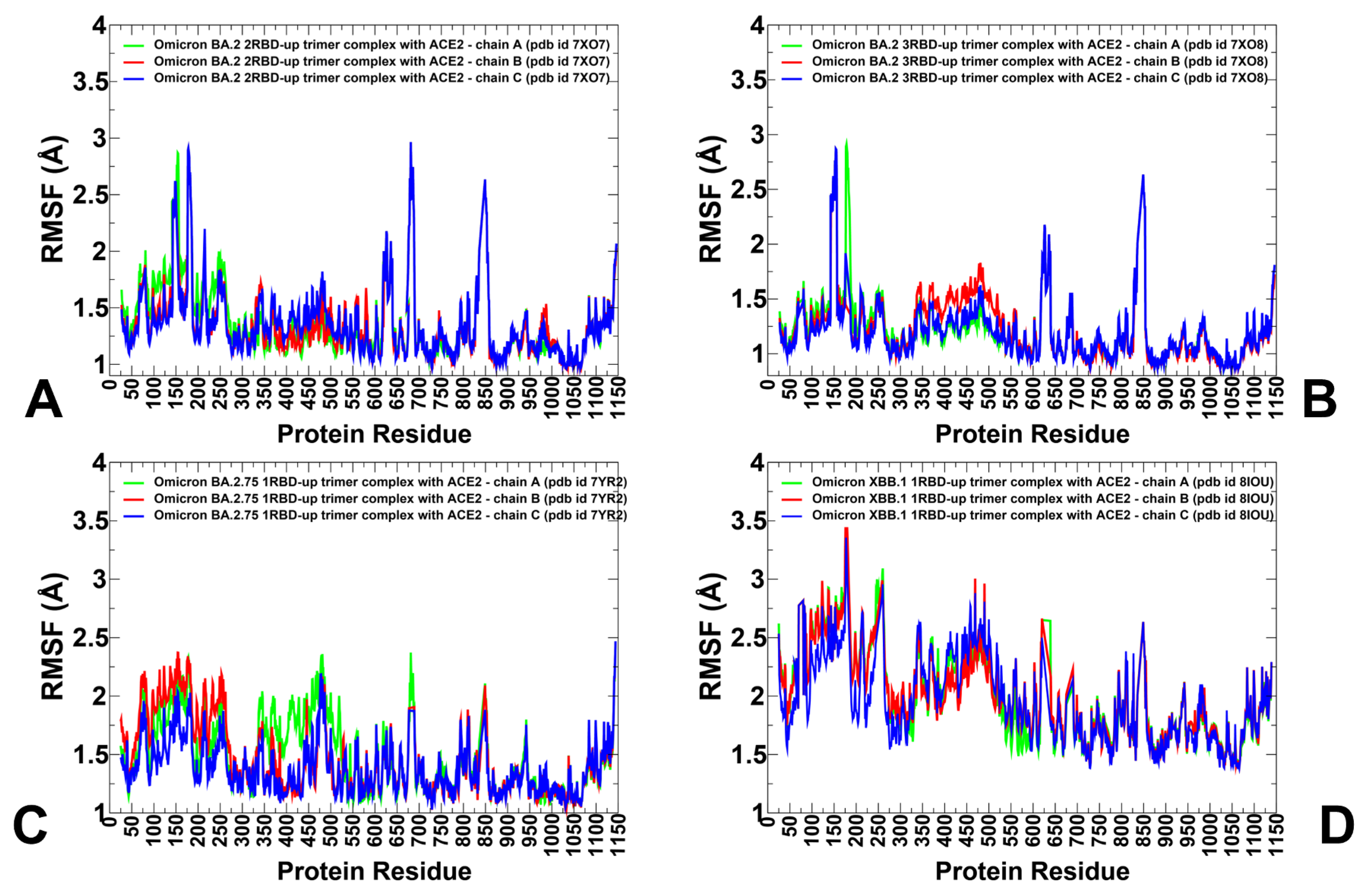
3.2. Coarse-Grained Simulations of the Omicron BA.2, BA.2.75 and XBB.1 S Trimer-ACE2 Complexes
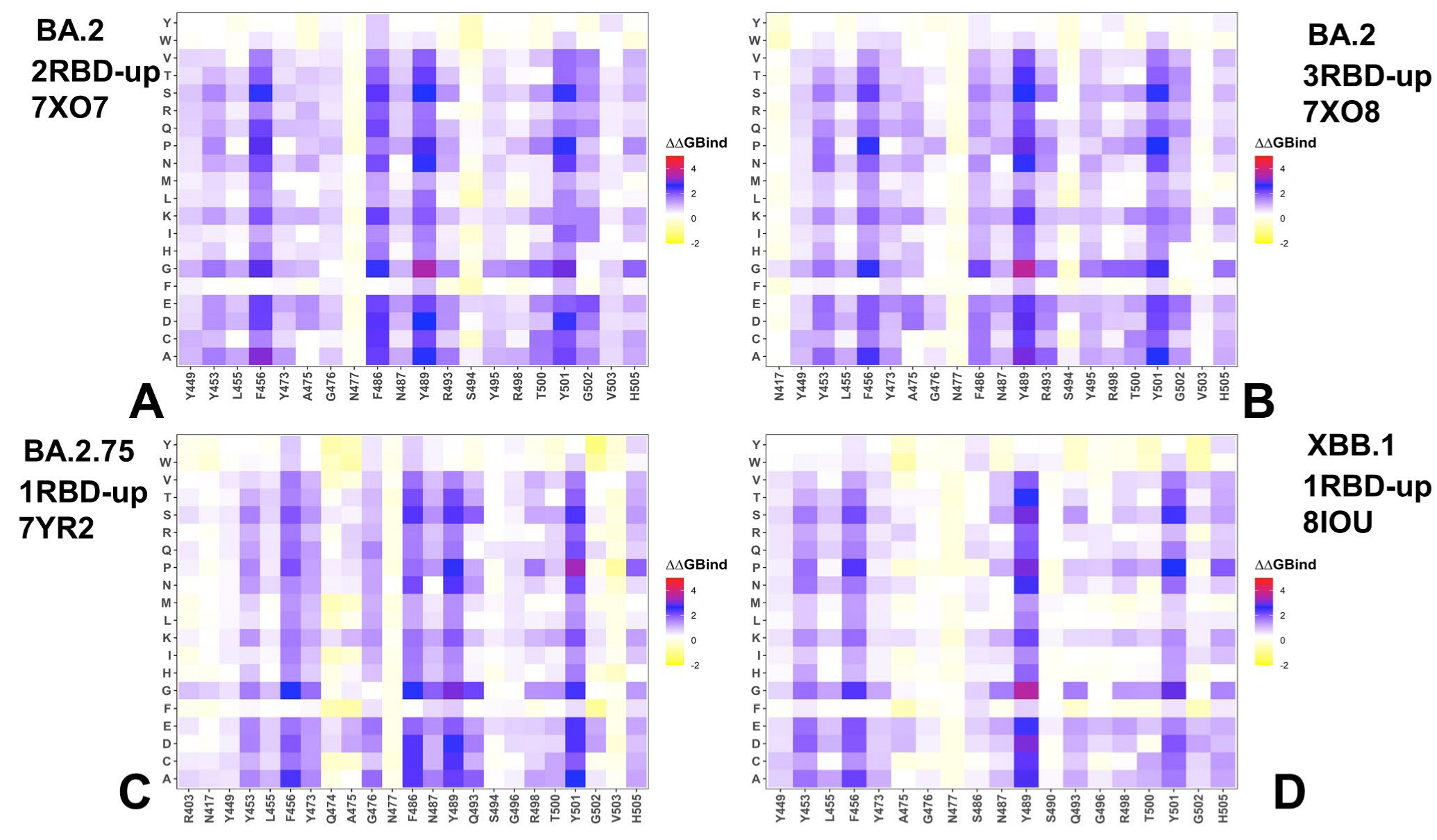
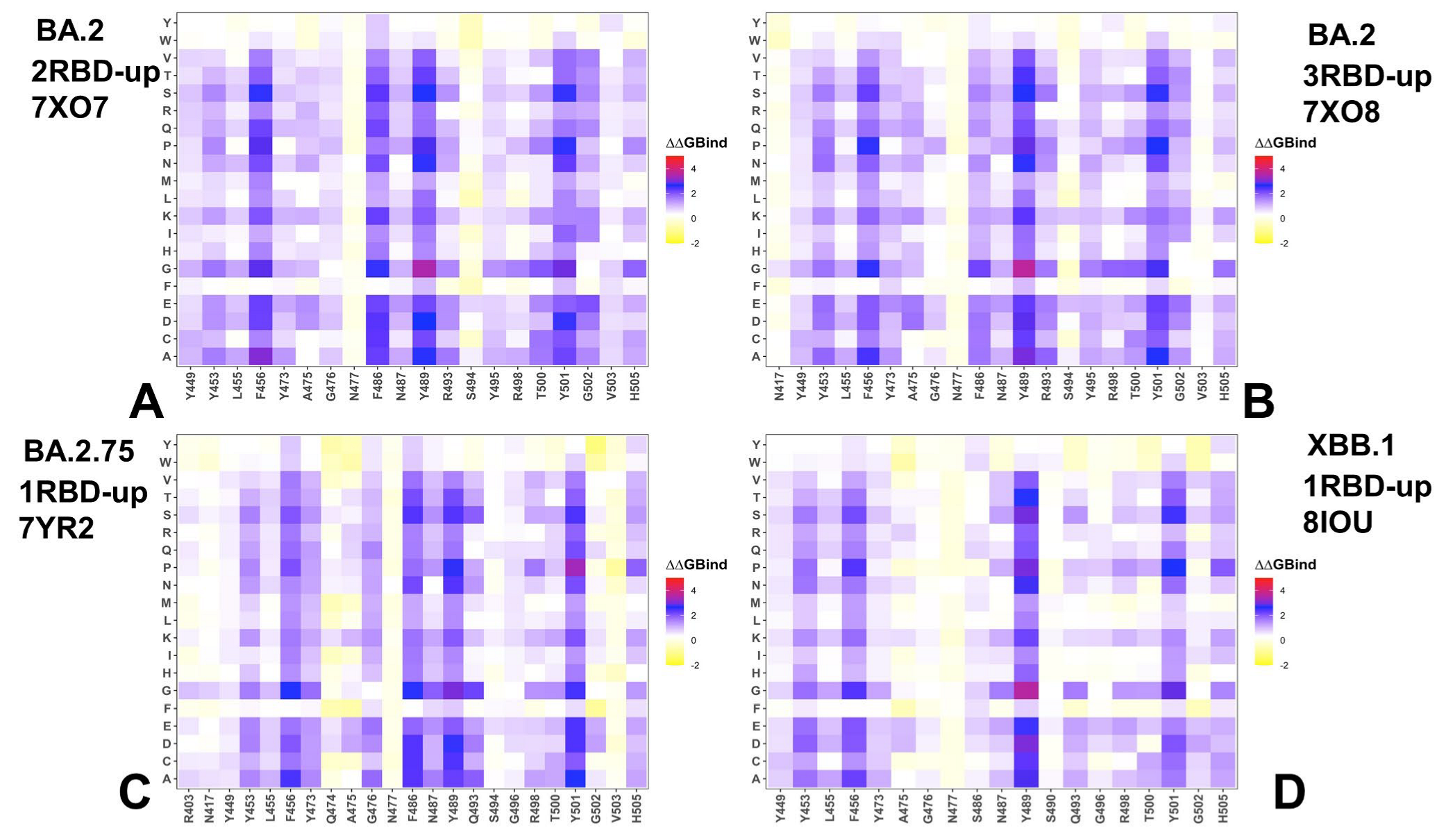
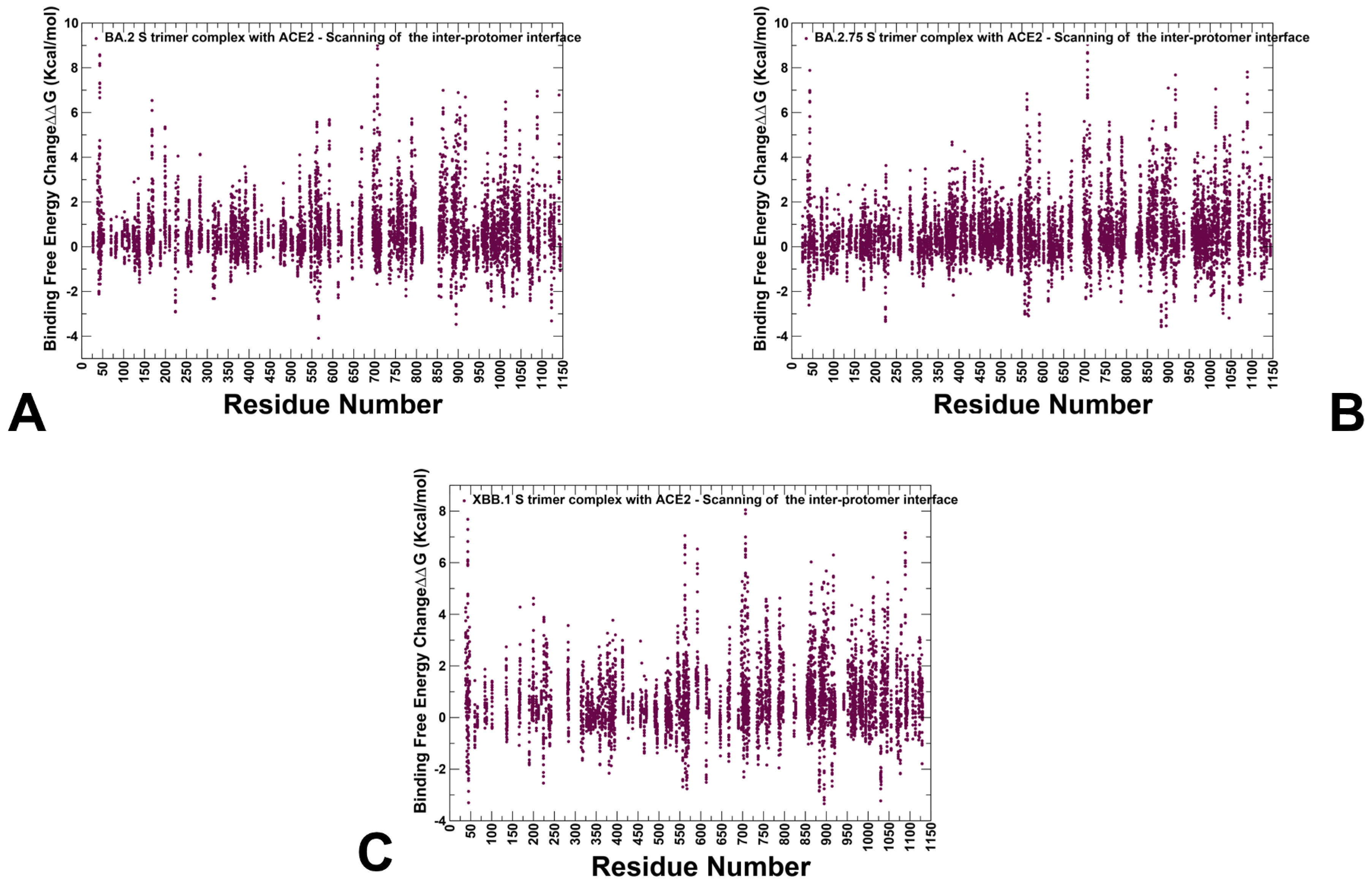
3.3. Mutational Scanning of the Binding and Inter-Protomer Interactions in the Omicron BA.2, BA.2.75 and XBB.1 S Trimer-ACE2 Complexes
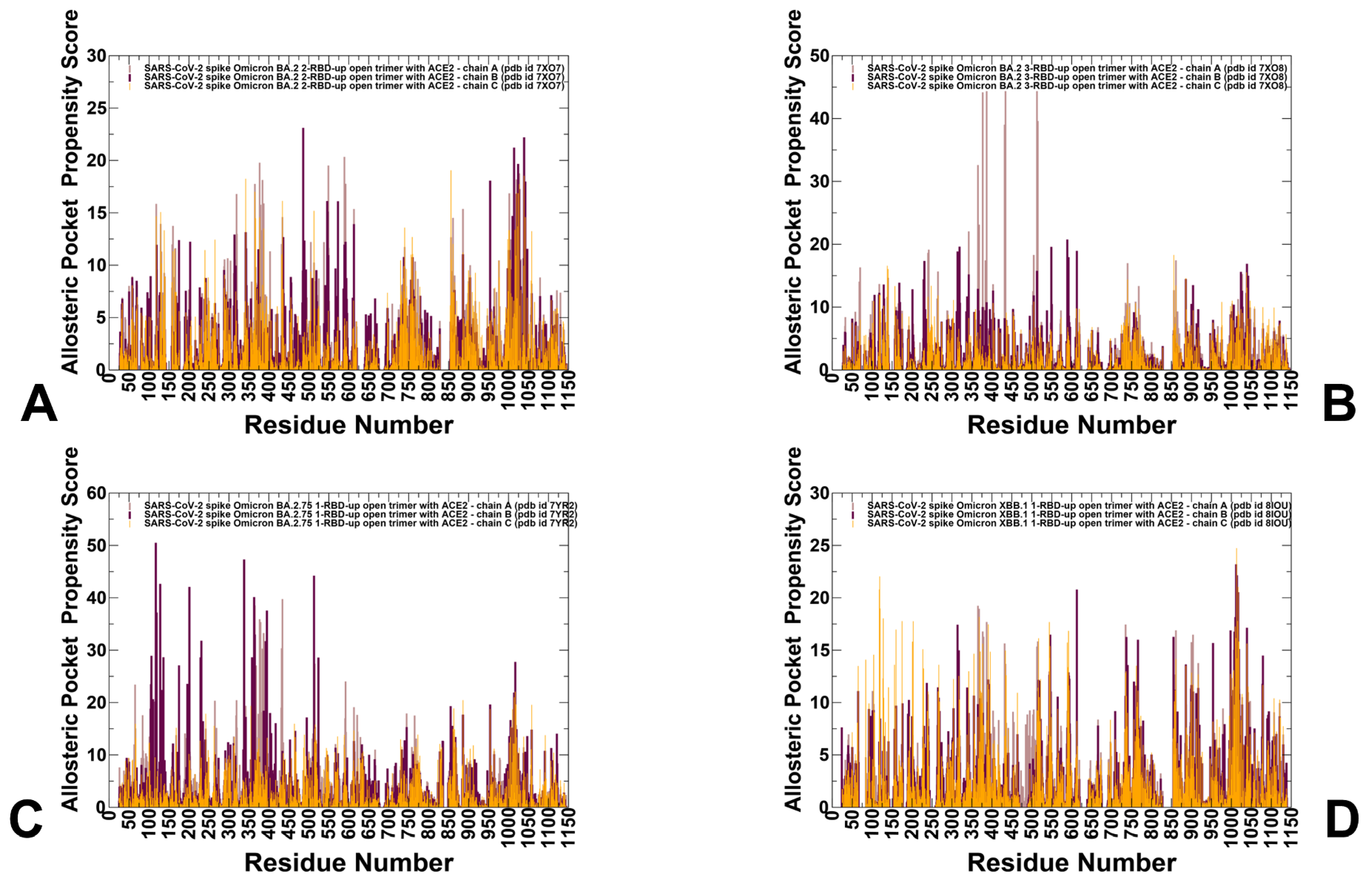
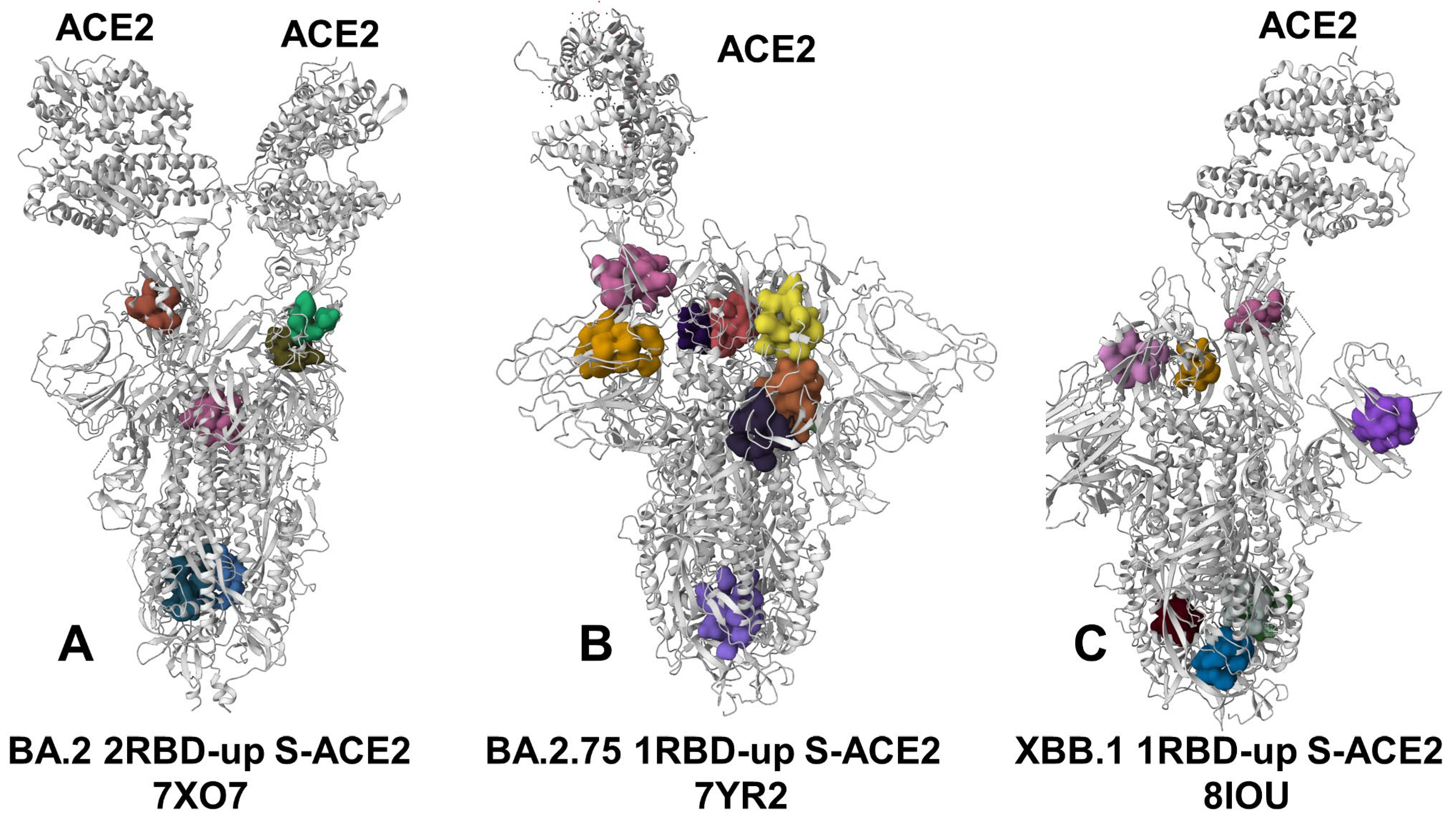
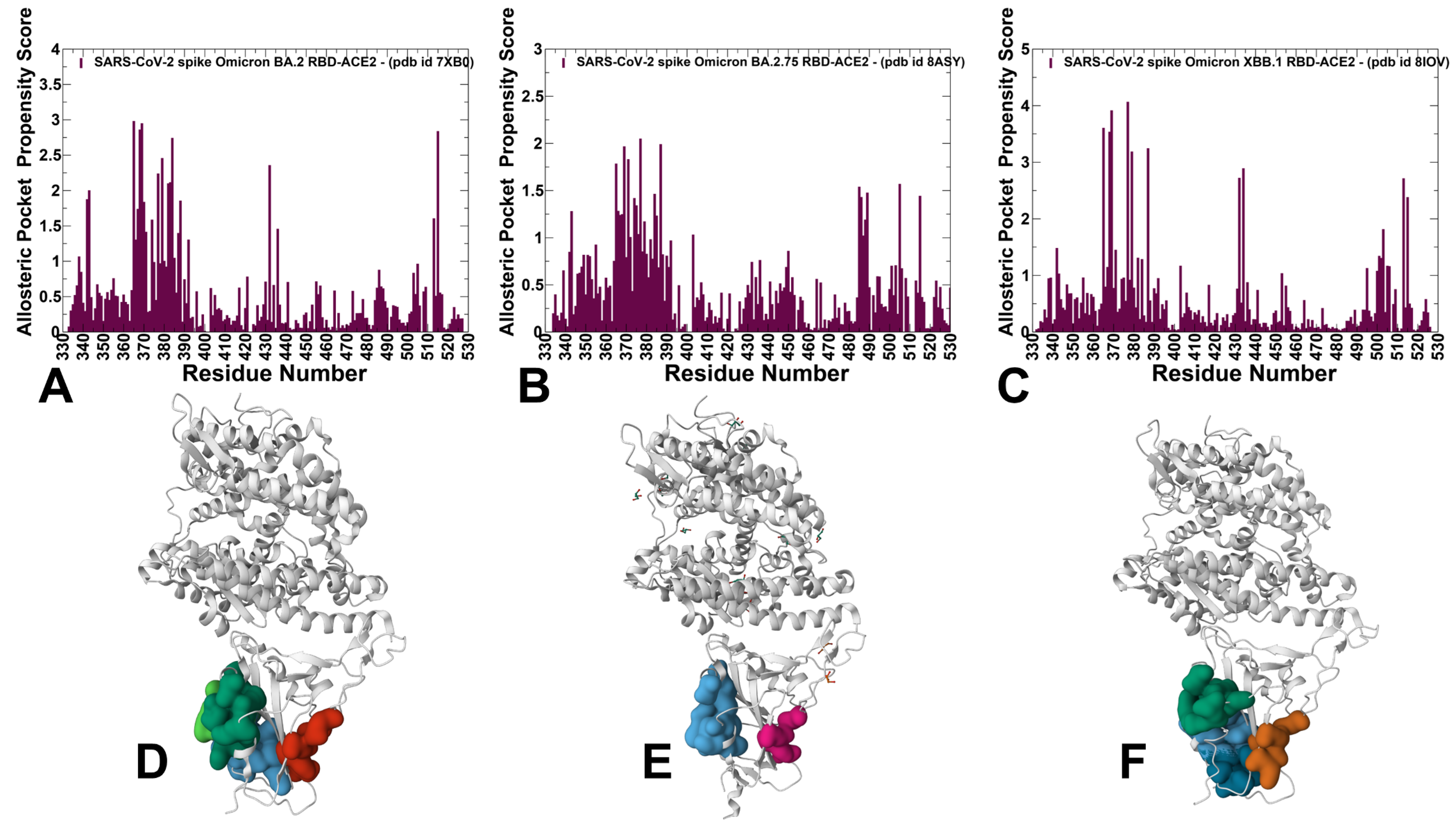
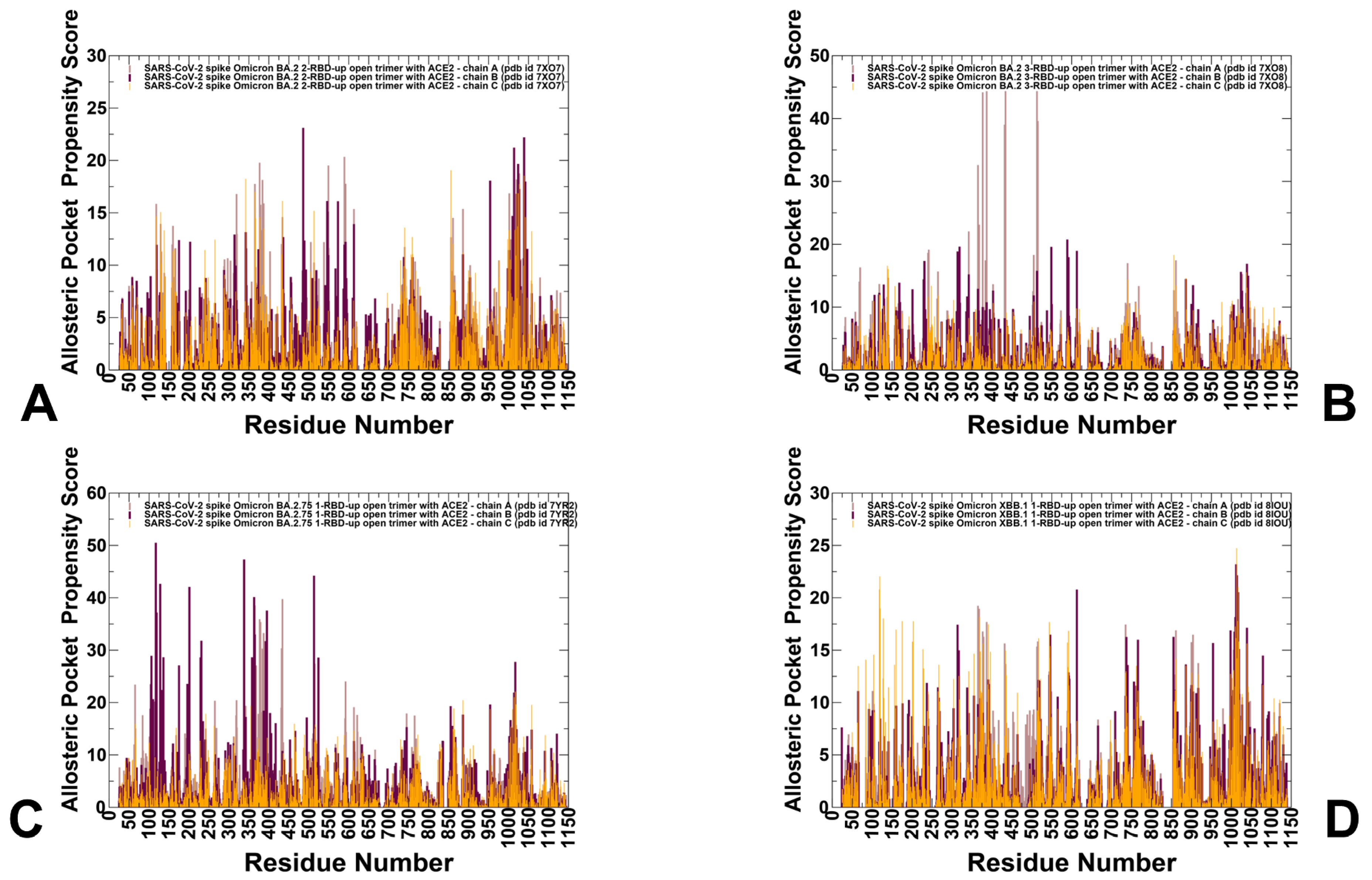
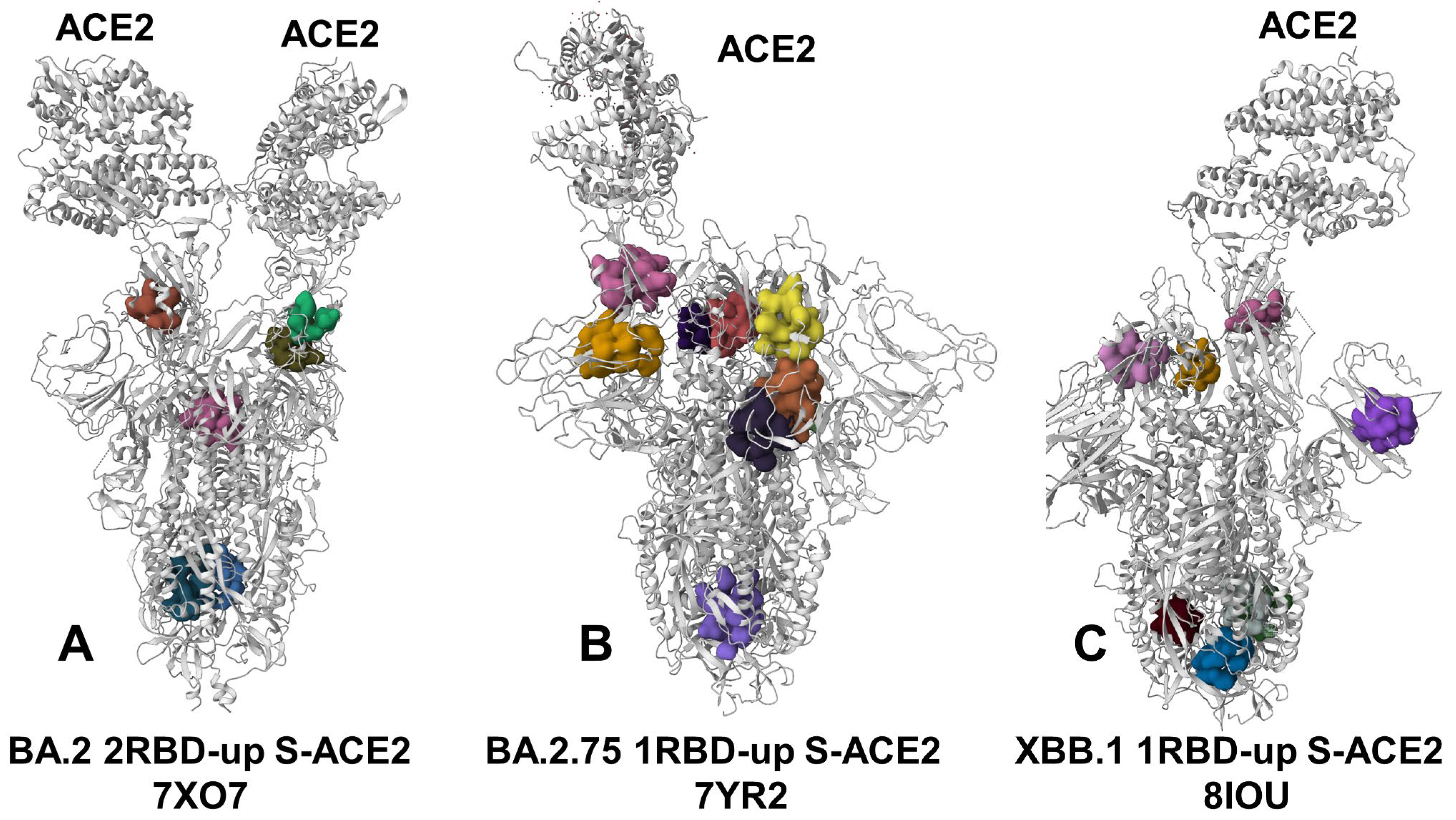
3.4. Detection of Cryptic Binding Pockets in the Conformational Ensembles of S Omicron Trimer Complexes with ACE2: The Effects of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe. 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. eLife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular basis of receptor binding and antibody neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural Diversity of the SARS-CoV-2 Omicron Spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wang, L.; Misasi, J.; Pegu, A.; Zhang, Y.; Harris, D.R.; Olia, A.S.; Talana, C.A.; Yang, E.S.; Chen, M.; et al. Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B.1.1.529. Science 2022, 376, eabn8897. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gao, Y.; Li, T.; Li, T.; Lu, Y.; Zheng, L.; Liu, Y.; Yang, T.; Luo, F.; Song, S.; et al. Structures of Omicron Spike Complexes and Implications for Neutralizing Antibody Development. Cell Rep. 2022, 39, 110770. [Google Scholar] [CrossRef] [PubMed]
- Stalls, V.; Lindenberger, J.; Gobeil, S.M.-C.; Henderson, R.; Parks, R.; Barr, M.; Deyton, M.; Martin, M.; Janowska, K.; Huang, X.; et al. Cryo-EM Structures of SARS-CoV-2 Omicron BA.2 Spike. Cell Rep. 2022, 39, 111009. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chen, Z.; Zhang, X.; Wen, A.; Yuan, X.; Yu, C.; Yang, J.; He, B.; Cao, Y.; Lu, G. Characterization of SARS-CoV-2 Omicron Spike RBD Reveals Significantly Decreased Stability, Severe Evasion of Neutralizing-Antibody Recognition but Unaffected Engagement by Decoy ACE2 Modified for Enhanced RBD Binding. Signal Transduct. Target. Ther. 2022, 7, 6. [Google Scholar] [CrossRef]
- Cerutti, G.; Guo, Y.; Liu, L.; Liu, L.; Zhang, Z.; Luo, Y.; Huang, Y.; Wang, H.H.; Ho, D.D.; Sheng, Z.; et al. Cryo-EM Structure of the SARS-CoV-2 Omicron Spike. Cell Rep. 2022, 38, 110428. [Google Scholar] [CrossRef]
- Ye, G.; Liu, B.; Li, F. Cryo-EM Structure of a SARS-CoV-2 Omicron Spike Protein Ectodomain. Nat Commun. 2022, 13, 1214. [Google Scholar] [CrossRef]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat. Commun. 2022, 13, 742. [Google Scholar] [CrossRef]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, C.; Cao, X.; Gu, C.; Liu, H.; Jiang, M.; Wang, X.; Yuan, Q.; Wu, K.; Liu, J.; et al. Structural and biochemical mechanism for increased infectivity and immune evasion of Omicron BA.2 variant compared to BA.1 and their possible mouse origins. Cell Res. 2022, 32, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tang, W.; Gao, H.; Lavine, C.L.; Shi, W.; Peng, H.; Zhu, H.; Anand, K.; Kosikova, M.; Kwon, H.J.; et al. Structural and Functional Characteristics of the SARS-CoV-2 Omicron Subvariant BA.2 Spike Protein. Nat. Struct. Mol. Biol. 2023, 30, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Song, W.; Wang, L.; Liu, P.; Yue, C.; Jian, F.; Yu, Y.; Yisimayi, A.; Wang, P.; Wang, Y.; et al. Characterization of the Enhanced Infectivity and Antibody Evasion of Omicron BA.2.75. Cell Host Microbe 2022, 30, 1527–1539.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, J.; Zheng, J.; Jin, Y.; Zhang, Y.; Tang, F.; Li, J.; Cheng, H.; Jiang, L.; Wen, H.; et al. Emerging Omicron Subvariants Evade Neutralizing Immunity Elicited by Vaccine or BA.1/BA.2 Infection. J. Med. Virol. 2023, 95, e28539. [Google Scholar] [CrossRef]
- Saito, A.; Tamura, T.; Zahradnik, J.; Deguchi, S.; Tabata, K.; Anraku, Y.; Kimura, I.; Ito, J.; Yamasoba, D.; Nasser, H.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2.75 Variant. Cell Host Microbe 2022, 30, 1540–1555.e15. [Google Scholar] [CrossRef]
- Qu, P.; Evans, J.P.; Zheng, Y.-M.; Carlin, C.; Saif, L.J.; Oltz, E.M.; Xu, K.; Gumina, R.J.; Liu, S.-L. Evasion of Neutralizing Antibody Responses by the SARS-CoV-2 BA.2.75 Variant. Cell Host Microbe 2022, 30, 1518–1526.e4. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming Antibody Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 Binding and Antibody Evasion in Enhanced Transmissibility of XBB.1.5. Lancet Infect. Dis. 2023, 23, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Nehlmeier, I.; Kempf, A.; Cossmann, A.; Schulz, S.R.; Morillas Ramos, G.; Manthey, L.A.; Jäck, H.-M.; Behrens, G.M.N.; et al. Profound Neutralization Evasion and Augmented Host Cell Entry Are Hallmarks of the Fast-Spreading SARS-CoV-2 Lineage XBB.1.5. Cell Mol. Immunol. 2023, 20, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.-L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 Spike Reversibly Samples an Open-Trimer Conformation Exposing Novel Epitopes. Nat. Struct. Mol. Biol. 2022, 29, 229–238. [Google Scholar] [CrossRef]
- Calvaresi, V.; Wrobel, A.G.; Toporowska, J.; Hammerschmid, D.; Doores, K.J.; Bradshaw, R.T.; Parsons, R.B.; Benton, D.J.; Roustan, C.; Reading, E.; et al. Structural Dynamics in the Evolution of SARS-CoV-2 Spike Glycoprotein. Nat. Commun. 2023, 14, 1421. [Google Scholar] [CrossRef]
- Braet, S.M.; Buckley, T.S.; Venkatakrishnan, V.; Dam, K.-M.A.; Bjorkman, P.J.; Anand, G.S. Timeline of Changes in Spike Conformational Dynamics in Emergent SARS-CoV-2 Variants Reveal Progressive Stabilization of Trimer Stalk with Altered NTD Dynamics. eLife 2023, 12, e82584. [Google Scholar] [CrossRef]
- Raghuvamsi, P.V.; Tulsian, N.K.; Samsudin, F.; Qian, X.; Purushotorman, K.; Yue, G.; Kozma, M.M.; Hwa, W.Y.; Lescar, J.; Bond, P.J.; et al. SARS-CoV-2 S Protein:ACE2 Interaction Reveals Novel Allosteric Targets. eLife 2021, 10, e63646. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, R.; Hodge, E.A.; Díaz-Salinas, M.A.; Nguyen, A.; Munro, J.B.; Lee, K.K. hACE2-Induced Allosteric Activation in SARS-CoV versus SARS-CoV-2 Spike Assemblies Revealed by Structural Dynamics. ACS Infect Dis. 2023, 9, 1180–1189. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free Fatty Acid Binding Pocket in the Locked Structure of SARS-CoV-2 Spike Protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Berger, I.; Schaffitzel, C. Cryo-EM Reveals Binding of Linoleic Acid to SARS-CoV-2 Spike Glycoprotein, Suggesting an Antiviral Treatment Strategy. Acta Crystallogr D Struct Biol. 2023, 79, 111–121. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Hodgson, L.; Williamson, M.K.; Buzas, D.; Borucu, U.; Powers, K.; Stenner, R.; Vasileiou, K.; et al. The Free Fatty Acid–Binding Pocket Is a Conserved Hallmark in Pathogenic β-Coronavirus Spike Proteins from SARS-CoV to Omicron. Sci. Adv. 2022, 8, eadc9179. [Google Scholar] [CrossRef] [PubMed]
- Hao, A.; Song, W.; Li, C.; Zhang, X.; Tu, C.; Wang, X.; Wang, P.; Wu, Y.; Ying, T.; Sun, L. Defining a Highly Conserved Cryptic Epitope for Antibody Recognition of SARS-CoV-2 Variants. Signal Transduct. Target. Ther. 2023, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.-H.; Portnoff, A.D.; et al. Structural Analysis of Full-Length SARS-CoV-2 Spike Protein from an Advanced Vaccine Candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; dos Santos, M.S.; et al. SARS-CoV-2 Can Recruit a Heme Metabolite to Evade Antibody Immunity. Sci. Adv. 2021, 7, eabg7607. [Google Scholar] [CrossRef]
- Altomare, C.G.; Adelsberg, D.C.; Carreno, J.M.; Sapse, I.A.; Amanat, F.; Ellebedy, A.H.; Simon, V.; Krammer, F.; Bajic, G. Structure of a Vaccine-Induced, Germline-Encoded Human Antibody Defines a Neutralizing Epitope on the SARS-CoV-2 Spike N-Terminal Domain. mBio 2022, 13, e0358021. [Google Scholar] [CrossRef]
- Silva, R.P.; Huang, Y.; Nguyen, A.W.; Hsieh, C.-L.; Olaluwoye, O.S.; Kaoud, T.S.; Wilen, R.E.; Qerqez, A.N.; Park, J.-G.; Khalil, A.M.; et al. Identification of a Conserved S2 Epitope Present on Spike Proteins from All Highly Pathogenic Coronaviruses. eLife 2023, 12, e83710. [Google Scholar] [CrossRef]
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface can Identify High-Affinity Variants Associated with Increased Transmissibility. J. Mol. Biol. 2021, 433, 167051. [Google Scholar] [CrossRef]
- Gan, H.H.; Zinno, J.; Piano, F.; Gunsalus, K.C. Omicron Spike Protein Has a Positive Electrostatic Surface That Promotes ACE2 Recognition and Antibody Escape. Front. Virol. 2022, 2, 894531. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; Giron, C.C.; Laaksonen, A. Electrostatic Features for the Receptor Binding Domain of SARS-CoV-2 Wildtype and Its Variants. Compass to the Severity of the Future Variants with the Charge-Rule. J. Phys. Chem. B. 2022, 126, 6835–6852. [Google Scholar] [CrossRef]
- Hristova, S.H.; Zhivkov, A.M. Omicron Coronavirus: pH-Dependent Electrostatic Potential and Energy of Association of Spike Protein to ACE2 Receptor. Viruses 2023, 15, 1752. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Giovanetti, M.; Benvenuto, D.; Angeletti, S.; Ceccarelli, G.; Pascarella, S.; Casu, M.; Fiori, P.L.; et al. On the SARS-CoV-2 BA.2.75 Variant: A Genetic and Structural Point of View. J. Med. Virol. 2023, 95, e28119. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Ciccozzi, M.; Bianchi, M.; Benvenuto, D.; Cauda, R.; Cassone, A. The Electrostatic Potential of the Omicron Variant Spike Is Higher than in Delta and Delta-plus Variants: A Hint to Higher Transmissibility? J. Med. Virol. 2022, 94, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Ciccozzi, M.; Bianchi, M.; Benvenuto, D.; Cauda, R.; Cassone, A. The Value of Electrostatic Potentials of the Spike Receptor Binding and N-Terminal Domains in Addressing Transmissibility and Infectivity of SARS-CoV-2 Variants of Concern. J Infect. 2022, 84, e62–e63. [Google Scholar] [CrossRef] [PubMed]
- Barre, A.; Klonjkowski, B.; Benoist, H.; Rougé, P. How Do Point Mutations Enhancing the Basic Character of the RBDs of SARS-CoV-2 Variants Affect Their Transmissibility and Infectivity Capacities? Viruses 2022, 14, 783. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G. Coarse-Grained Molecular Simulations and Ensemble-Based Mutational Profiling of Protein Stability in the Different Functional Forms of the SARS-CoV-2 Spike Trimers: Balancing Stability and Adaptability in BA.1, BA.2 and BA.2.75 Variants. Int. J. Mol. Sci. 2023, 24, 6642. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.J.; Chang, Y.C.; Ko, T.P.; Draczkowski, P.; Chien, Y.C.; Wu, K.P.; Khoo, K.H.; Chang, H.W.; Hsu, S.D. Cryo-EM analysis of a feline coronavirus spike protein reveals a unique structure and camouflaging glycans. Proc. Natl. Acad. Sci. USA 2020, 117, 1438–1446. [Google Scholar] [CrossRef]
- Watanabe, Y.; Berndsen, Z.T.; Raghwani, J.; Seabright, G.E.; Allen, J.D.; Pybus, O.G.; McLellan, J.S.; Wilson, I.A.; Bowden, T.A.; Ward, A.B.; et al. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020, 11, 2688. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Harbison, A.M.; Fogarty, C.A.; Phung, T.K.; Satheesan, A.; Schulz, B.L.; Fadda, E. Fine-Tuning the Spike: Role of the Nature and Topology of the Glycan Shield in the Structure and Dynamics of the SARS-CoV-2 S. Chem. Sci. 2022, 13, 386–395. [Google Scholar] [CrossRef]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-receptor interactions of glycosylated SARS-CoV-2 spike and human ACE2 receptor. Cell Host Microbe 2020, 28, 586–601. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond shielding: The roles of glycans in the SARS-CoV-2 spike potein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Barros, E.P.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Wang, Y.; Fallon, L.; Raguette, L.; Belfon, K.; Simmerling, C.; Amaro, R.E. The Flexibility of ACE2 in the Context of SARS-CoV-2 Infection. Biophys J. 2021, 120, 1072–1084. [Google Scholar] [CrossRef]
- Mehdipour, A.R.; Hummer, G. Dual Nature of Human ACE2 Glycosylation in Binding to SARS-CoV-2 Spike. Proc. Natl. Acad. Sci. USA 2021, 118, e2100425118. [Google Scholar] [CrossRef]
- von Bülow, S.; Sikora, M.; Blanc, F.E.C.; Covino, R.; Hummer, G. Antibody accessibility determines location of spike surface mutations in SARS-CoV-2 variants. PLoS Comput. Biol. 2023, 19, e1010822. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 Spike Opening Dynamics and Energetics Reveal the Individual Roles of Glycans and Their Collective Impact. Commun. Biol. 2022, 5, 1170. [Google Scholar] [CrossRef]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of Interactions Regulating Conformational Stability and Dynamics of SARS-CoV-2 S-Protein. Biophys J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef]
- Zuzic, L.; Samsudin, F.; Shivgan, A.T.; Raghuvamsi, P.V.; Marzinek, J.K.; Boags, A.; Pedebos, C.; Tulsian, N.K.; Warwicker, J.; MacAry, P.; et al. Uncovering Cryptic Pockets in the SARS-CoV-2 Spike Glycoprotein. Structure 2022, 30, 1062–1074.e4. [Google Scholar] [CrossRef]
- Ghoula, M.; Naceri, S.; Sitruk, S.; Flatters, D.; Moroy, G.; Camproux, A.C. Identifying Promising Druggable Binding Sites and Their Flexibility to Target the Receptor-Binding Domain of SARS-CoV-2 Spike Protein. Comput. Struct. Biotechnol. J. 2023, 21, 2339–2351. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets. Int. J. Mol. Sci. 2022, 23, 11542. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Zhang, Y.; Zhang, X.; Zhang, L.; Shang, W.; Bai, F. Probing the Allosteric Inhibition Mechanism of a Spike Protein Using Molecular Dynamics Simulations and Active Compound Identifications. J. Med. Chem. 2021, 65, 2827–2835. [Google Scholar] [CrossRef]
- Li, B.; Wang, L.; Ge, H.; Zhang, X.; Ren, P.; Guo, Y.; Chen, W.; Li, J.; Zhu, W.; Chen, W.; et al. Identification of Potential Binding Sites of Sialic Acids on the RBD Domain of SARS-CoV-2 Spike Protein. Front. Chem. 2021, 9, 659764. [Google Scholar] [CrossRef]
- Guo, L.; Lin, S.; Chen, Z.; Cao, Y.; He, B.; Lu, G. Targetable Elements in SARS-CoV-2 S2 Subunit for the Design of Pan-Coronavirus Fusion Inhibitors and Vaccines. Signal Transduct. Target. Ther. 2023, 8, 197. [Google Scholar] [CrossRef]
- Xing, L.; Xu, X.; Xu, W.; Liu, Z.; Shen, X.; Zhou, J.; Xu, L.; Pu, J.; Yang, C.; Huang, Y.; et al. A Five-Helix-Based SARS-CoV-2 Fusion Inhibitor Targeting Heptad Repeat 2 Domain against SARS-CoV-2 and Its Variants of Concern. Viruses 2022, 14, 597. [Google Scholar] [CrossRef]
- Ma, J.; Su, D.; Sun, Y.; Huang, X.; Liang, Y.; Fang, L.; Ma, Y.; Li, W.; Liang, P.; Zheng, S. Cryo-Electron Microscopy Structure of S-Trimer, a Subunit Vaccine Candidate for COVID-19. J Virol. 2021, 95, e00194-21. [Google Scholar] [CrossRef]
- Huo, J.; Dijokaite-Guraliuc, A.; Liu, C.; Zhou, D.; Ginn, H.M.; Das, R.; Supasa, P.; Selvaraj, M.; Nutalai, R.; Tuekprakhon, A.; et al. A Delicate Balance between Antibody Evasion and ACE2 Affinity for Omicron BA.2.75. Cell Rep. 2023, 42, 111903. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A template based loop structure prediction server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An Interactive Web Server for Efficient Protein Structure Refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles. J. Phys. Chem. B. 2018, 122, 6673–6689. [Google Scholar] [CrossRef]
- Braun, E.; Gilmer, J.; Mayes, H.B.; Mobley, D.L.; Monroe, J.I.; Prasad, S.; Zuckerman, D.M. Best Practices for Foundations in Molecular Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5957. [Google Scholar] [CrossRef]
- Ke, Q.; Gong, X.; Liao, S.; Duan, C.; Li, L. Effects of Thermostats/Barostats on Physical Properties of Liquids by Molecular Dynamics Simulations. J. Mol. Liq. 2022, 365, 120116. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Haque, I.S.; Beauchamp, K.A.; Pande, V.S. A Fast 3 × N Matrix Multiply Routine for Calculation of Protein RMSD. bioRxiv 2014. [Google Scholar] [CrossRef]
- Theobald, D.L. Rapid Calculation of RMSDs Using a Quaternion-Based Characteristic Polynomial. Acta Crystallogr. A. 2005, 61, 478–480. [Google Scholar] [CrossRef]
- Liu, P.; Agrafiotis, D.K.; Theobald, D.L. Fast Determination of the Optimal Rotational Matrix for Macromolecular Superpositions. J. Comput. Chem. 2010, 31, 1561–1563. [Google Scholar] [CrossRef]
- Naritomi, Y.; Fuchigami, S. Slow Dynamics in Protein Fluctuations Revealed by Time-Structure Based Independent Component Analysis: The Case of Domain Motions. J. Chem. Phys. 2011, 134, 065101. [Google Scholar] [CrossRef]
- Schwantes, C.R.; Pande, V.S. Improvements in Markov State Model Construction Reveal Many Non-Native Interactions in the Folding of NTL9. J. Chem. Theory Comput. 2013, 9, 2000–2009. [Google Scholar] [CrossRef]
- Sultan, M.M.; Pande, V.S. tICA-Metadynamics: Accelerating Metadynamics by Using Kinetically Selected Collective Variables. J. Chem. Theory Comput. 2017, 13, 2440–2447. [Google Scholar] [CrossRef]
- Trozzi, F.; Wang, X.; Tao, P. UMAP as a Dimensionality Reduction Tool for Molecular Dynamics Simulations of Biomacromolecules: A Comparison Study. J. Phys. Chem. B. 2021, 125, 5022–5034. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef]
- Wu, H.; Paul, F.; Wehmeyer, C.; Noe, F. Multiensemble Markov models of molecular thermodynamics and kinetics. Proc. Natl. Acad. Sci. USA 2016, 113, E3221–E3230. [Google Scholar] [CrossRef]
- Suárez, E.; Adelman, J.L.; Zuckerman, D.M. Accurate Estimation of Protein Folding and Unfolding Times: Beyond Markov State Models. J. Chem. Theory Comput. 2016, 12, 3473–3481. [Google Scholar] [CrossRef]
- Bowman, G.R.; Bolin, E.R.; Hart, K.M.; Maguire, B.C.; Marqusee, S. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. USA 2015, 112, 2734–2739. [Google Scholar] [CrossRef]
- Bowman, G.R.; Noé, F. Software for Building Markov State Models. Adv. Exp. Med. Biol. 2014, 797, 139. [Google Scholar] [CrossRef]
- Bowman, G.R. A Tutorial on Building Markov State Models with MSMBuilder and Coarse-Graining Them with BACE. Methods Mol Biol. 2014, 1084, 141–158. [Google Scholar] [CrossRef]
- Trendelkamp-Schroer, B.; Wu, H.; Paul, F.; Noé, F. Estimation and Uncertainty of Reversible Markov Models. J. Chem. Phys. 2015, 143, 174101. [Google Scholar] [CrossRef]
- Bowman, G.R.; Huang, X.; Pande, V.S. Using Generalized Ensemble Simulations and Markov State Models to Identify Conformational States. Methods 2009, 49, 197–201. [Google Scholar] [CrossRef]
- Sacquin-Mora, S.; Lavery, R. Investigating the local flexibility of functional residues in hemoproteins. Biophys. J. 2006, 90, 2706–2717. [Google Scholar] [CrossRef]
- Sacquin-Mora, S.; Laforet, E.; Lavery, R. Locating the active sites of enzymes using mechanical properties. Proteins 2007, 67, 350–359. [Google Scholar] [CrossRef]
- Sacquin-Mora, S. Bridging Enzymatic Structure Function via Mechanics: A Coarse-Grain Approach. Methods Enzymol. 2016, 578, 227–248. [Google Scholar] [CrossRef]
- Ermak, D.L.; McCammon, J.A. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978, 69, 1352–1360. [Google Scholar] [CrossRef]
- Pastor, R.W.; Venable, R.; Karplus, M. Brownian dynamics simulation of a lipid chain in a membrane bilayer. J. Chem. Phys. 1988, 89, 1112–1127. [Google Scholar] [CrossRef]
- Rotkiewicz, P.; Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.E.; Marti, M.A.; Capece, L. CG2AA: Backmapping protein coarse-grained structures. Bioinformatics 2016, 32, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef]
- Dehouck, Y.; Gilis, D.; Rooman, M. A new generation of statistical potentials for proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef]
- Krivák, R.; Hoksza, D. P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J. Cheminform. 2018, 10, 39. [Google Scholar] [CrossRef]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated Ligand-Binding Site Predictions for Experimental and Modelled Protein Structures. Nucleic Acids Res. 2022, 50, W593–W597. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Ghersi, D.; Sanchez, R. EasyMIFS and SiteHound: A toolkit for the identification of ligand-binding sites in protein structures. Bioinformatics 2009, 25, 3185–3186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of Cavities on Protein Surface Using Multiple Computational Approaches for Drug Binding Site Prediction. Bioinformatics 2011, 27, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Tian, H.; Tao, P. PASSer2.0: Accurate Prediction of Protein Allosteric Sites Through Automated Machine Learning. Front. Mol. Biosci. 2022, 9, 879251. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Xiao, S.; Jiang, X.; Tao, P. PASSer: Fast and Accurate Prediction of Protein Allosteric Sites. Nucleic Acids Res. 2023, 51, W427–W431. [Google Scholar] [CrossRef]
- Tian, H.; Xiao, S.; Jiang, X.; Tao, P. PASSerRank: Prediction of allosteric sites with learning to rank. J. Comput. Chem. 2023, 44, 2223–2229. [Google Scholar] [CrossRef]
- Trigueiro-Louro, J.; Correia, V.; Figueiredo-Nunes, I.; Gíria, M.; Rebelo-de-Andrade, H. Unlocking COVID Therapeutic Targets: A Structure-Based Rationale against SARS-CoV-2, SARS-CoV and MERS-CoV Spike. Comput. Struct. Biotechnol. J. 2020, 18, 2117–2131. [Google Scholar] [CrossRef]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchianò, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 Receptor Interaction by Natural and Semi-Synthetic Steroidal Agents Acting on Functional Pockets on the Receptor Binding Domain. Front Chem. 2020, 8, 572885. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A Web Server for Automatic Binding Site Prediction, Analysis and Druggability Assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef]
- Borrel, A.; Regad, L.; Xhaard, H.; Petitjean, M.; Camproux, A.-C. PockDrug: A Model for Predicting Pocket Druggability That Overcomes Pocket Estimation Uncertainties. J. Chem. Inf. Model. 2015, 55, 882–895. [Google Scholar] [CrossRef]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.-C. PockDrug-Server: A New Web Server for Predicting Pocket Druggability on Holo and Apo Proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G. Balancing Functional Tradeoffs between Protein Stability and ACE2 Binding in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB Lineages: Dynamics-Based Network Models Reveal Epistatic Effects Modulating Compensatory Dynamic and Energetic Changes. Viruses 2023, 15, 1143. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Stewart, C.M.; Walls, A.C.; Hannon, W.W.; Veesler, D.; Bloom, J.D. Deep Mutational Scans for ACE2 Binding, RBD Expression, and Antibody Escape in the SARS-CoV-2 Omicron BA.1 and BA.2 Receptor-Binding Domains. PLoS Pathog. 2022, 18, e1010951. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Iketani, S.; Li, Z.; Guo, Y.; Yeh, A.Y.; Liu, M.; Yu, J.; Sheng, Z.; Huang, Y.; Liu, L.; et al. Antigenic Characterization of the SARS-CoV-2 Omicron Subvariant BA.2.75. Cell Host Microbe 2022, 30, 1512–1517.e4. [Google Scholar] [CrossRef]
- Zhao, Z.; Xie, Y.; Bai, B.; Luo, C.; Zhou, J.; Li, W.; Meng, Y.; Li, L.; Li, D.; Li, X.; et al. Structural Basis for Receptor Binding and Broader Interspecies Receptor Recognition of Currently Circulating Omicron Sub-Variants. Nat Commun. 2023, 14, 4405. [Google Scholar] [CrossRef]
- Qing, E.; Li, P.; Cooper, L.; Schulz, S.; Jäck, H.-M.; Rong, L.; Perlman, S.; Gallagher, T. Inter-Domain Communication in SARS-CoV-2 Spike Proteins Controls Protease-Triggered Cell Entry. Cell Rep. 2022, 39, 110786. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion Mechanism of 2019-nCoV and Fusion Inhibitors Targeting HR1 Domain in Spike Protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (Previously 2019-nCoV) Infection by a Highly Potent Pan-Coronavirus Fusion Inhibitor Targeting Its Spike Protein That Harbors a High Capacity to Mediate Membrane Fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J. Phys. Chem. B 2021, 125, 850–873. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omicron Variant | Mutational Landscape |
|---|---|
| BA.1 | A67, T95I, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F |
| BA.2 | T19I, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| BA.2.75 | T19I, G142D, K147E, W152R, F157L, I210V, V213G, G257S, G339H, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, G446N, N460K, S477N, T478K, E484A, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| XBB.1 | T19I, V83A, G142D, Del144, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486S, F490S, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P.; Verkhivker, G. Comparative Analysis of Conformational Dynamics and Systematic Characterization of Cryptic Pockets in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB.1 Spike Complexes with the ACE2 Host Receptor: Confluence of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites. Viruses 2023, 15, 2073. https://doi.org/10.3390/v15102073
Alshahrani M, Gupta G, Xiao S, Tao P, Verkhivker G. Comparative Analysis of Conformational Dynamics and Systematic Characterization of Cryptic Pockets in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB.1 Spike Complexes with the ACE2 Host Receptor: Confluence of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites. Viruses. 2023; 15(10):2073. https://doi.org/10.3390/v15102073
Chicago/Turabian StyleAlshahrani, Mohammed, Grace Gupta, Sian Xiao, Peng Tao, and Gennady Verkhivker. 2023. "Comparative Analysis of Conformational Dynamics and Systematic Characterization of Cryptic Pockets in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB.1 Spike Complexes with the ACE2 Host Receptor: Confluence of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites" Viruses 15, no. 10: 2073. https://doi.org/10.3390/v15102073
APA StyleAlshahrani, M., Gupta, G., Xiao, S., Tao, P., & Verkhivker, G. (2023). Comparative Analysis of Conformational Dynamics and Systematic Characterization of Cryptic Pockets in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB.1 Spike Complexes with the ACE2 Host Receptor: Confluence of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites. Viruses, 15(10), 2073. https://doi.org/10.3390/v15102073